Org. Synth. 1985, 63, 109
DOI: 10.15227/orgsyn.063.0109
DIASTEREOSELECTIVE α-ALKYLATION OF β-HYDROXYCARBOXYLIC ESTERS THROUGH ALKOXIDE ENOLATES: DIETHYL (2S, 3R)-(+)-3-ALLYL-2-HYDROXYSUCCINATE FROM DIETHYL (S)-( − )-MALATE
[Butanedioic acid, 2-hydroxy-3-(2-propenyl)-, diethyl ester, [S-(R,S)]]
Submitted by Dieter Seebach, Johannes Aebi, and Daniel Wasmuth
1.
Checked by Brian Maxwell and Clayton H. Heathcock.
1. Procedure
A
500-mL, three-necked flask containing a
magnetic stirring bar is equipped with a
100-mL pressure-equalizing and serum-capped dropping funnel, a
three-way stopcock, and a
low-temperature thermometer (Note 1). The dry apparatus is filled with
argon and kept under an inert gas pressure of ca. 100 mm against the atmosphere until the aqueous workup
(Note 2); see
Figure 1.
Figure 1
The flask is charged through serum cap B with 17 mL (120 mmol) of diisopropylamine (Note 3) and 200 mL of tetrahydrofuran (THF) (Note 4), using syringe techniques. It is cooled to −75°C in a dry ice bath. With stirring, exactly 100 mmol of butyllithium (hexane solution) (Note 5) is introduced from the dropping funnel (Note 6) within 10 min, followed after 0.5 hr, by a mixture of 9.51 g (50 mmol) of ( − )-diethyl (S)-malate (Note 7) and 5 mL of THF, which is added dropwise through cap B at such a rate that the temperature does not rise above −60°C. The addition takes approximately 10 min (Note 8). The dry ice cooling bath is replaced by an ice–salt bath (ca. −15°C) in which the contents of the flask warm to −20°C within 0.5 hr. The solution is stirred at −20°C ± 2°C for 0.5 hr and then is cooled to −75°C.
To the solution of the alkoxide enolate thus prepared is added by syringe within 5 min 10.7 mL (124 mmol) of neat 3-bromo-1-propene (Note 9) at such a rate that the temperature of the reaction mixture does not rise above −70°C. Stirring is continued, first for 2 hr at −75°C, and then overnight while the temperature rises to −5°C (Note 10).
The reaction mixture is quenched by adding a solution of
12 g (200 mmol) of glacial acetic acid in 20 mL of diethyl ether at −50°C and is then poured into a
1-L separatory funnel containing
500 mL of ether and 70 mL of water. The organic layer is washed successively with
40 mL each of saturated sodium bicarbonate and sodium chloride solution, and the aqueous phases are extracted with two
200-mL portions of ether. The combined ethereal solutions are dried by vigorous stirring with dry
MgSO4 for 15 min. Removal of the solvent first with a
rotary evaporator at a bath temperature no higher than 35°C and then at room temperature under
oil pump vacuum (0.1 mm) furnishes
10.4 g of a yellow oil consisting, according to capillary gas chromatography (GC)
(Note 11), of 81.3% of the desired allylated (2S,3R) product (
73.5% yield), 8.5% of the (2S,3S) diastereoisomer (90.5% ds
2), and
6.3% of the starting diethyl malate (Note 12).
The product is purified by flash chromatography (Note 13),(Note 14),(Note 15). A flash column of 7-cm diameter is charged with 450 g of silica gel (Kieselgel 60, Merck, Korngrösse 0.040–0.063 mm, 230–400 mesh ASTM) and 10.4 g of the crude product. A 1 : 1 mixture of ether and pentane is used for elution, with a running rate of 5-cm column length per minute (pressure 1.25 atm). After a 200-mL forerun, 33-mL fractions are collected. No attempt is made to separate the two diastereoisomers; fractions 22–40 are combined to give 8.0 g (70%) of pure allylated product [ratio of diastereoisomers 92 : 8 (Note 11)], after removal of the solvent; [α]D20 + 11.2° (chloroform, c 2.23) (Note 16).
2. Notes
1.
A
Pt-100 thermometer (Testoterm KG, Lenzkirch, Germany) was used by the submitters. This is preferred to a conventional thermometer because it is more accurate and more convenient to read. Careful temperature control is essential for the present procedure. Unless stated otherwise, all temperatures given are those of the reaction mixture. The checkers found that a
+30 to −100°C alcohol thermometer is satisfactory.
2.
The glass components of the apparatus are dried overnight in a 170°C
oven and allowed to cool in a
desiccator over a drying agent before assembly. The apparatus is filled with
argon by evacuating and pressurizing several times through the three-way stopcock, as described previously.
33.
Diisopropylamine was freshly distilled from
calcium hydride.
4.
Tetrahydrofuran (THF) was first distilled under an inert atmosphere from KOH and then from the blue solution obtained with
potassium and
benzophenone, as described previously.
3 [However, see warning notice,
Org. Synth., Coll. Vol. V 1973, 976–977.]
5.
Before use, the commercial
1.6 M solution of butyllithium in hexane was titrated acidimetrically using
diphenylacetic acid as an indicator.
46.
The dropping funnel was calibrated before use in this procedure. With standard
graduated dropping funnels and syringes, the submitters noticed up to 10% deviation from true volumes! Syringe techniques were applied; the dropping funnel was rinsed with ca.
5 mL of dry THF.
7.
Commercial
(S)-( − )-malic acid was esterified under standard conditions, following a procedure by Fischer and Speier.
5 The freshly distilled ester employed by the submitters had an
[α]D20 −10.5° (neat) (
d204 = 1.128 g/cm
3), which corresponds to an optical purity of 100%.
68.
The flask, in which the ester/THF mixture was prepared, and the syringe are rinsed with a total of ca.
5 mL of dry THF.
9.
Commercial
allyl bromide was distilled before use.
10.
The submitters used a
2-L Dewar cylinder holding, besides the flask, ca.
1 L of ethanol as a cooling liquid. If no excess dry ice was present at the beginning of the warm-up period, it took ca. 12 hr to reach −5°C.
11.
GLC-analysis were performed using the following column and conditions: 0.3-mm ×
20-m glass capillary column Pluronic L 64, program 120°C, (3 min), 10°C/min up to 200°C, temperature of injector and detector 200°C, carrier gas:
hydrogen (1.3 atm).
12.
A total of ca. 4% of four minor side products with longer retention times is also present.
13.
This is the fastest method, although it consumes large amounts of solvent and of silica gel. The procedure is that of Still et al.
7 Conventional chromatography is also possible but is more time-consuming.
14.
Kugelrohr distillation does not separate the starting material,
diethyl malate. Distillation through a
30-cm Vigreux column (silvered vacuum jacket) leads to loss of material (only
40% yield, diastereoisomer ratio 90 : 10, free of starting material).
15.
Hydrolysis of the crude product yields, after recrystallization, pure
(2S,3R)-3-allyl-2-hydroxysuccinic acid, mp
96.0–97.5°C,
[α]D20 +14.7° (
acetone,
c 1.69).
16.
The boiling point is
77–78°C (0.07 mm). Previously, a specific rotation of
[α]D25 + 11.9° (
chloroform,
c 1.77) was reported.
8 The
13C NMR spectrum (CDCl
3) of the (2
S, 3
R) isomer shows the following signals δ (off-resonance multiplicity, assignment): 14.12 (q, CO
2CH
2CH
3), 32.21 (t, C(3)
CH
2), 48.25 [d, C(3)], 60.86 and 61.81 (2 t, CO
2CH
2CH
3), 70.36 [d, C(2)], 117.78 [t,
C(3)CH
2CH=
CH
2], 134.94 [d, C(3)CH
2CH=CH
2], 171.92 and 173.48 (2 s,
CO
2CH
2CH
3).
3. Discussion
The compound described here had not been known prior to our first synthesis of it.
8 9 10 Generally, aldol derivatives of this configuration are prepared by the addition of E enolates of esters to aldehydes,
11,12 1 →
2 in Scheme 1.
The method of preparing α-branched β-hydroxy esters by alkylation of dianion derivatives of the parent compounds was first discovered by Herrmann and Schlessinger.
13 It is highly diastereoselective
14 and applicable without racemization to optically active derivatives, as first demonstrated independently by Fráter with
β-hydroxybutanoate15 and by us with malate
8 9 10,16 17 18 (see
3 →
2 and
3 →
5 in Scheme 1). In the meantime, many applications have been published.
19 20,21 A related method of preparing derivatives belonging to the same diastereoisomeric series is the alkylation of β-lactone enolates.
22Examples of alkylation of malic esters are listed in Table I, together with those of double alkylation, which can also be achieved, see
2 →
4 in Scheme 1. Since the
(S) and the (R) forms of malic acid are both readily available,
23 the enantiomers of all structures shown in Table I can be prepared as well. The method is also applicable to β-hydroxy γ-lactones of type
6, the alkylations of which lead
24 25 26 to derivatives of opposite configuration
8, see
6 →
7 in Scheme 2. Finally, the dilithio derivative
9 of di-
tert-butyl
N-formylaspartate is alkylated (→
10; see Scheme 2)
27 with the same relative topicity,
28 ul, as the malate dianion derivative (Table I).
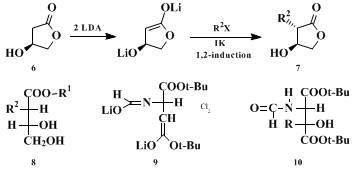
TABLE I
PRODUCTS OF MONO- AND DIALKYLATION WITH RELATIVE TOPICITY ula OF (S)-MALIC ESTERS THROUGH ALKOXIDE ENOLATES. THE RATIOS OF DIASTEREOISOMERS (SEE % ds) WAS DETERMINED BY 1H or 13C NMR SPECTROSCOPY OR BY GC ANALYSIS.
|
|
|
|
Product
|
R1
|
R2
|
R3
|
Yield (%)
|
% dsb
|
Ref.
|
|
|
|
|
|
|
(Malate → a)
|
|
a
|
CH3
|
CH3
|
—
|
65
|
91
|
8,17
|
|
|
CH3
|
C(OH)(CH3)2
|
—
|
55
|
75
|
8
|
|
|
C2H5
|
CH3
|
—
|
88
|
91
|
8
|
|
|
C2H5
|
CH2C6H5
|
—
|
48
|
91
|
8
|
|
|
C2H5
|
I
|
—
|
80
|
67
|
8
|
|
|
CH3
|
CH2CH2NO2
|
—
|
31
|
85
|
16
|
|
|
CH3
|
C2H5
|
—
|
64
|
90
|
17
|
|
|
CH3
|
CH2CH=CH2
|
—
|
63
|
93
|
8b,14c
|
|
|
|
|
|
(a → b)
|
|
b
|
CH3
|
CH3
|
CH3
|
94
|
—
|
17
|
|
|
CH3
|
CH3
|
C2H5
|
36
|
95
|
17
|
|
|
CH3
|
C2H5
|
CH3
|
|
72
|
17
|
|
|
CH3
|
CH3
|
CD3
|
92
|
89
|
17
|
|
|
CH3
|
CH3
|
13CH3
|
81
|
88
|
17
|
|
|
CH3
|
CH3
|
CH2CH=CH2
|
74
|
95
|
18
|
|
|
CH3
|
CH3
|
H
|
>98
|
67
|
18
|
|
|
|
|
|
In Table II, a series of useful chiral building blocks is shown, which are accessible through alkylations of malic acid derivatives; the table also contains some natural products that were synthesized from such building blocks.
The alkylation of doubly deprotonated β-hydroxy esters, an example of which is described in the procedure above, is not just a useful alternative to the diastereoselective aldol-type addition, but can supply enantiomerically pure products from appropriate precursors, and it can be used for the preparation of α,α-disubstituted derivatives (see
4 in Scheme 1). These were hitherto not available stereoselectively from enolates of α-branched esters and aldehydes.
29,30
This preparation is referenced from:
TABLE II
CHIRAL, NONRACEMIC BUILDING BLOCKS AND NATURAL PRODUCTS SYNTHESIZED THROUGH ALKYLATION OF MALIC ACID DERIVATIVESa
|
|
Products and Intermediates from (S)-Malic Acid
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
aThe four-carbon unit of the structure derived from malic acid is indicated by heavy lines.
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
allylated product
(S) and the (R) forms of malic acid
ethanol (64-17-5)
acetic acid (64-19-7)
ether,
diethyl ether (60-29-7)
hydrogen (1333-74-0)
chloroform (67-66-3)
sodium bicarbonate (144-55-8)
sodium chloride (7647-14-5)
Allyl bromide,
3-bromo-1-propene (106-95-6)
acetone (67-64-1)
Benzophenone (119-61-9)
potassium (7440-09-7)
Diphenylacetic acid (117-34-0)
Pentane (109-66-0)
MgSO4 (7487-88-9)
malic acid,
(617-48-1)
malate
butyllithium (109-72-8)
Tetrahydrofuran,
THF (109-99-9)
hexane (110-54-3)
argon (7440-37-1)
calcium hydride (7789-78-8)
diisopropylamine (108-18-9)
diethyl malate,
,
(626-11-9)
β-hydroxybutanoate
DIETHYL (2S, 3R)-(+)-3-ALLYL-2-HYDROXYSUCCINATE (73837-97-5)
Butanedioic acid, 2-hydroxy-3-(2-propenyl)-, diethyl ester, [S-(R,S)]
(2S,3R)-3-allyl-2-hydroxysuccinic acid
(S)-Malic Acid (97-67-6)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved