Org. Synth. 1993, 71, 63
DOI: 10.15227/orgsyn.071.0063
STEREOCONTROLLED PREPARATION OF 3-ACYLTETRAHYDROFURANS FROM ACID-PROMOTED REARRANGEMENTS OF ALLYLIC KETALS: (2S,3S)-3-ACETYL-8-CARBOETHOXY-2,3-DIMETHYL-1-OXA-8-AZASPIRO[4.5]DECANE
Submitted by Larry E. Overman and Gilbert M. Rishton
1.
Checked by Takashi Ooi and Hisashi Yamamoto.
1. Procedure
Caution! tert-Butylithium is extremely pyrophoric and must not be allowed to come into contact with the atmosphere. This reagent should only be handled by individuals trained in its proper and safe use. It is recommended that transfers be carried out by using a 20-mL or smaller glass syringe filled to no more than 2/3 capacity, or by cannula. For a discussion of procedures for handling air-sensitive reagents, see Aldrich Technical Bulletin AL-134. [Note added August 2009]
A.
(2R,3S)- and (2S,3S)-1,4-Dioxa-2,3-dimethyl-2-(1-methylethenyl)-8-carboethoxy-8-azaspiro[4.5]decane. An
oven-dried, 500-mL, three-necked, round-bottomed flask is fitted with a
mechanical stirrer,
100-mL addition funnel, and
rubber septum, and then is charged with
100 mL of dry tetrahydrofuran (Note 1) and
7.7 mL (10.5 g, 86.7 mmol) of 2-bromopropene (Note 2). The solution is cooled to −70°C with mechanical stirring and a
1.9 M pentane solution of tert-butyllithium (92 mL, 175 mmol) is added by syringe over 20 min. The resulting yellow solution is stirred for an additional 10 min at −70°C and at this time a solution of
17.6 g (54.0 mmol) of 3-(S)-[(tert-butyldiphenylsilyl)oxy]-2-butanone2 and
50 mL of dry tetrahydrofuran is added by
dropping funnel over 20 min. The resulting solution is stirred for an additional 30 min at −70°C and at this time a
1.0 M tetrahydrofuran solution of tetrabutylammonium fluoride (163 mL, 163 mmol) is added in one portion and the resulting mixture is warmed to 23°C and stirred for 1 hr. At this time the contents of the flask are poured into
200 mL of saturated aqueous ammonium chloride (NH
4Cl) and the resulting mixture is concentrated to remove
tetrahydrofuran. The resulting aqueous suspension is diluted with
200 mL of brine and extracted twice with
200 mL of ethyl acetate (Note 3). The combined organic extracts are washed with five
100-mL portions of brine, dried over
sodium sulfate, filtered, and then concentrated under reduced pressure using a
rotary evaporator. The residue is subjected to short path vacuum distillation (150–160°C, 3 mm) to remove the less volatile
tert-butyldiphenylsilyl by-product. The distillate contains ca.
10 g of a colorless oil that is comprised of the 2,3-dimethyl-1-pentene-3,4-diols as a 6:1 mixture of diastereomers and up to
30% of tributylamine (Note 4) and
(Note 5).
1-Carbethoxy-4-piperidone (7.52 g, 43.9 mmol) (Note 6) and p-toluenesulfonic acid (5.0 g, 26 mmol) are added to a 250-mL, round-bottomed flask that contains the above distillate and a magnetic stir bar. The mixture is stirred under vacuum (20 mm) at 100°C for 90 min and the evolved water vapor is collected in a vacuum trap. The mixture is cooled to 23°C and subjected to flash chromatography on silica gel (250 g, 20 cm × 10 cm) using ethyl acetate:hexane (1:4) as the eluant (Note 7) to give 9.0 g (59% overall) of (2R,3S)- and (2S,3S)-1,4-dioxa-2,3-dimethyl-2-(1-methylethenyl)-8-carboethoxy-8-azaspiro[4.5]decane, a 6:1 mixture of diastereomers, as a pale yellow oil (Note 8).
B. (2S,3S)-3-Acetyl-8-carboethoxy-2,3-dimethyl-1-oxa-8-azaspiro[4.5]decane. Dry nitromethane (100 mL) (Note 9) is added through a rubber septum by syringe to a vacuum-dried, 500-mL, round-bottomed flask that contains the ketal mixture prepared in Step A (9.00 g, 31.8 mmol) and a magnetic stir bar. The solution is cooled to −23°C, tin(IV) chloride (SnCl4) (11 mL, 94 mmol) is added by syringe and the solution is stirred for 30 min at −23°C (Note 10). At this time the brown solution is warmed to 23°C and stirring is continued for an additional 30 min. Saturated aqueous NH4Cl (200 mL) is added and the mixture is concentrated under reduced pressure using a rotary evaporator to remove nitromethane. The resulting aqueous suspension is extracted with ethyl acetate (200 mL) and the organic extract is washed with brine (200 mL), dried over sodium sulfate (Na2SO4) and concentrated under reduced pressure using a rotary evaporator. The residue is subjected to flash chromatography on silica gel (250 g, 20 cm × 10 cm) using ethyl acetate:hexane (1:1) eluant (Note 7) to give 8.1 g (90%) of (2S,3S)-3-acetyl-8-carboethoxy-2,3-dimethyl-1-oxa-8-azaspiro[4.5]decane as a pale yellow oil (Note 11) and (Note 12).
2. Notes
1.
Anhydrous
tetrahydrofuran was prepared by distillation under
argon from
sodium benzophenone ketyl.
2.
2-Bromopropene, obtained from Aldrich Chemical Company, Inc., was distilled and then passed through a plug of activity IV basic alumina immediately before use.
3.
The fine white emulsion formed at this stage was collected with the organic phase and was cleared in the subsequent
brine washings.
4.
This crude material was acceptable for use in the second step, although more
p-toluenesulfonic acid will be required if large amounts of
tributylamine are present. The diol mixture, free from
tributylamine, can be obtained by careful chromatography on silica gel using
ethyl acetate-hexane (1:1). The purified sample has the following characteristics:
1H NMR (500 MHz, CDCl
3, major isomer) δ: 1.10 (d, 3 H, J = 6.5, CH
3), 1.37 (s, 3 H, CH
3), 1.80 (s, 3 H, CH
3), 2.21 (br s, 2 H, 2 × OH), 3.77 (q, 1 H, J = 5.6, CH), 4.89 (d, 1 H, J = 1.1, CH=C), 5.06 (s, 1 H, CH=C); IR (film) cm
−1: 3421, 3397, 3390, 3364, 2981, 2937, 1088; MS (Cl) m/z 113.0936 (113.0966 calcd for C
7H
14O
2, MH – H
2O).
5.
The major isomer is assigned the 3R, 4S stereochemistry on the expectation that the addition would occur preferentially with Cram (Felkin-Ahn) selectivity.
3 This assignment was confirmed by
1H NMR DNOE experiments on the
isobutyraldehyde acetal.
6.
1-Carbethoxy-4-piperidone was obtained from Aldrich Chemical Company, Inc., and used as received.
7.
A series of 200-mL fractions was collected during flash chromatography. The product was eluted in fractions 3–8 as indicated by TLC analysis using
4% ethanolic phosphomolybdic acid stain.
8.
This sample has the following characteristics:
1H NMR (500 MHz, CDCl
3, major isomer) δ: 1.17 (d, 3 H, J = 5.1, CH
3), 1.26 (t, 3 H, J = 7.1, OCH
2CH
3), 1.45 (s, 3 H, CH
3), 1.77 (s, 3 H, CH
3C=), 1.60–1.81 (m, 5 H, 2 × CH
2 and CH), 3.43–3.75 (m, 4 H, 2 × CH
2N), 4.13 (q, 2 H, J = 7.1, OCH
2CH
3), 4.96 (s, 2 H, CH
2=C); IR (film) cm
−1: 2977, 1702, 1433, 1238, 1122; MS (Cl) m/z 284.1850 (284.1861 calcd for C
15H
25NO
4, MH). Anal. Calcd for C
15H
25NO
4: C, 63.58; H, 8.89; N, 4.94. Found: C, 63.48; H, 8.90; N, 4.89.
9.
Nitromethane was dried by distillation of a 10:1 mixture of
nitromethane and
trifluoroacetic anhydride and collection of the center fraction that distilled at
100°C.
10.
Tin(IV) chloride (SnCl4) was obtained from Aldrich Chemical Company, Inc., and handled under an atmosphere of
argon.
11.
Gas chromatographic analysis using a
25-m 10% SP 2100 silicone column showed that this sample was 94% pure and contained one major unidentified impurity. Bulb-to-bulb distillation (200°C, 0.6 mm) of a
7.4-g sample of the crude product afforded
7.0 g (
85%) of the product as a pale yellow oil, which was shown by GLC analysis to be of 100% purity. This sample has the following spectral characteristics: [α]
D −79.1° (MeOH,
c 1.0);
1H NMR (500 MHz, CDCl
3) δ: 1.17 (d, 3 H, J = 6.6, CH
3), 1.25 (m, 6 H, OCH
2CH
3 and CH
3), 1.70–1.90 (m, 4 H, 2 × CH
2), 2.19 (s, 3 H, CH
3CO), 1.57 (d, 1 H, J = 13.5) 2.36 (d, 1 H, J = 13.5), 3.38–3.70 (m, 4 H, 2 × CH
2N), 3.89 (q, 1 H, J = 6.6, CH) 4.12 (q, 2 H, J = 7.1, OCH
2CH
3);
13C NMR (125 MHz, CDCl
3) δ: 14.5, 15.6, 22.5, 28.3, 36.0, 37.0, 40.7, 41.1, 47.3, 58.4, 61.0, 79.1, 81.0, 155.5, 210.3; IR (film) cm
−1: 2977, 2937, 1705, 1701, 1698, 1472, 1455, 1434, 1365, 1356, 1274, 1237; MS (Cl) m/z 284.1845 (284.1860 calcd for C
15H
25NO
4, MH). Anal. Calcd for C
15H
25NO
4: C, 63.58; H, 8.89; N, 4.94. Found: C, 63.38; H, 8.87; N, 4.88.
12.
The enantiomeric excess of the product is >96%. This was determined by treating a sample of the ketone with
sodium borohydride/
methanol (NaBH
4/MeOH) (23°C) and separating the resulting 3:2 mixture of alcohol diastereomers by flash chromatography (silica gel, 2:3
ethyl acetate-hexane). The major alcohol diastereomer was converted to its Mosher ester
4 [2.5 eq of
(+)-α-methoxytrifluoromethylphenylacetic acid, 3 eq of
dicyclohexylcarbodiimide, and 0.2 eq of
4-(dimethylamino)pyridine, CH
2Cl
2] and the crude esterification reaction mixture was analyzed using 500 MHz
1H NMR. None of the minor diastereomer was observed while doping experiments established that 2% would have been detected [diagnostic signals: δ 1.80 (δ, J = 13.4, major ester diastereomer); δ 1.82 (δ, J = 14.1, minor ester diastereomer)].
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
This procedure illustrates a fundamentally new method for constructing substituted tetrahydrofurans.
5,6,7,8,9,10 This practical method assembles the
tetrahydrofuran ring from allylic diol and carbonyl components and in the process forms three ring bonds: C(2)-C(3), C(4)-C(5) and O-C(5). Both aldehydes (eq 1) and ketones (illustrated in the present procedure) can be employed as the carbonyl component. Although it is often convenient to isolate the acetal intermediate, conversion to the
3-acyltetrahydrofuran can also be accomplished in many cases by the direct reaction of the diol and carbonyl components.
8 High cis stereoselectivity (at least 20:1) is observed in the preparation of tetrahydrofurans that contain single side chains at carbons 2 and 5 (eq. 1). The kinetically controlled product also has the cis relationship of these side chains and the 3-acyl substituent.
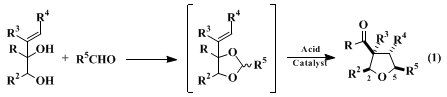
A definitive feature of this highly stereoselective new route to substituted tetrahydrofurans is that both syn and anti allylic diol stereoisomers typically afford identical
tetrahydrofuran products. Thus, there is no need for stereoselective construction of the allylic diol reaction partner. The construction of substituted tetrahydrofurans in high enantiomeric purity from non-racemic allylic diol precursors has also been established.
5,7 The rearrangement illustrated in eq. 2 is the key step in a recent synthesis of
(+)-muscarine.
The scope and mechanism of the SnCl
4-promoted rearrangement of allylic acetals have been investigated in detail and these studies provide considerable guidance for using this new
tetrahydrofuran synthesis.
5,6,7,8,9 Three major limitations emerge from studies conducted to date: (1) When the
tetrahydrofuran construction involves a ketone, and thus forms a quaternary center at C(5), allylic diols with alkene substituents more nucleophilic than terminal vinyl rearrange in highest yield. (2) Allylic acetals that are reluctant to ring open in the presence of acid catalysts to generate oxocarbenium ions often undergo decomposition, rather than conversion to acyltetrahydrofuran products. (3) Allylic acetals that form highly stabilized oxocarbeniums (e.g., cinnamaldehyde-derived acetals) do not undergo conversion to 3-acyltetrahydrofurans.
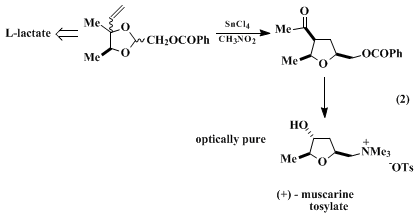
This procedure illustrates the asymmetric synthesis of a spirobicyclic
tetrahydrofuran from the reaction of readily available
(S)-3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-butanone2 with cyclic ketones. The specific example described here affords an azaspirobicyclic
tetrahydrofuran 1 that is structurally related to a recently reported class of powerful muscarinic agonists, exemplified by
2.
10 Consistent with limitation (1) noted above, the related reaction of
3-methyl-4-pentene-2,3-diol (which contains a less-nucleophilic terminal vinyl participant) occurs in lower yield. As with other acetals that contain an electron-withdrawing heteroatom β or γ to the
acetal carbon, the rearrangement described in this procedure is more efficient in
nitromethane than in the less-ionizing solvent
dichloromethane (CH
2Cl
2).
7This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
brine
sodium benzophenone ketyl
(2R,3S)- and (2S,3S)-1,4-Dioxa-2,3-dimethyl-2-(1-methylethenyl)-8-carboethoxy-8-azaspiro[4.5]decane
2,3-dimethyl-1-pentene-3,4-diols
ethanolic phosphomolybdic acid
3-acyltetrahydrofuran
(+)-muscarine
ethyl acetate (141-78-6)
methanol (67-56-1)
ammonium chloride (12125-02-9)
sodium sulfate (7757-82-6)
Pentane (109-66-0)
Nitromethane (75-52-5)
dichloromethane (75-09-2)
tin(IV) chloride (7646-78-8)
Tetrahydrofuran (109-99-9)
hexane (110-54-3)
argon (7440-37-1)
sodium borohydride (16940-66-2)
dicyclohexylcarbodiimide (538-75-0)
tributylamine (102-82-9)
trifluoroacetic anhydride (407-25-0)
p-toluenesulfonic acid (104-15-4)
ethyl acetate-hexane (2639-63-6)
acetal carbon (463-57-0)
Tetrabutylammonium fluoride (429-41-4)
2-Bromopropene (557-93-7)
4-(dimethylamino)pyridine (1122-58-3)
tert-Butyllithium (594-19-4)
(+)-α-methoxytrifluoromethylphenylacetic acid (56135-03-6)
(2S,3S)-3-Acetyl-8-carboethoxy-2,3-dimethyl-1-oxa-8-azaspiro[4.5]decane (155534-75-1)
3-(S)-[(tert-Butyldiphenylsilyl)oxy]-2-butanone,
(S)-3-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-2-butanone (135367-18-9)
tert-butyldiphenylsilyl
1-Carbethoxy-4-piperidone (29976-53-2)
isobutyraldehyde acetal
3-methyl-4-pentene-2,3-diol
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved