Due to their importance in, for instance, natural products,
2 bioactive molecules,
3 pharmaceuticals,
4 nutrients (vitamins),
5 agrochemicals,
6 dyes,
7 liquid crystals,
8 and functional polymers,
9 heteroaryl units have been recognized as essential structural motifs in various realms. The significance has motivated organic chemists to develop new methods and strategies for more efficiently constructing bonds on heteroaryl scaffolds. One of the most frequently utilized strategies for this purpose is transition metal catalysis, where diverse types of bonds can now be constructed on heteroaryl rings.
10 Another option for functionalizing heteroaryl rings is the nucleophilic aromatic substitution (S
NAr) reaction; however, this reaction has often been unsatisfactory. Despite its long history of use, the negative impression seems to be ascribed to critical limitations. Aromatic compounds are intrinsically electron-rich due to their (4n + 2)π electrons but must react with electron-rich nucleophiles in the S
NAr process (Scheme 1a). This demand has narrowed the scope of substrates. Thus, anionic nucleophiles with highly electropositive metals and/or electron-poor heteroaryl electrophiles with one or more electron-withdrawing groups have been utilized (Scheme 1a).
11 This substrate combination appears most frequently in the conventional S
NAr reaction via an addition-elimination sequence where a Meisenheimer intermediate is involved. This mechanism can be viewed as the cause of the negative image of the conventional S
NAr reaction. However, the appearance of the concerted S
NAr reaction has triggered a major breakthrough.
12 The key feature thereof is that heteroaryl electrophiles without EWGs can serve as substrates, while metal nucleophiles are still needed in most cases (Scheme 1b).
12,13 The expanded scope of the heteroaryl electrophile is due presumably to an alternate mechanism involving a single transition state that does not require the disruption of aromaticity by way of the Meisenheimer intermediate, thereby lowering the activation energy of the process.
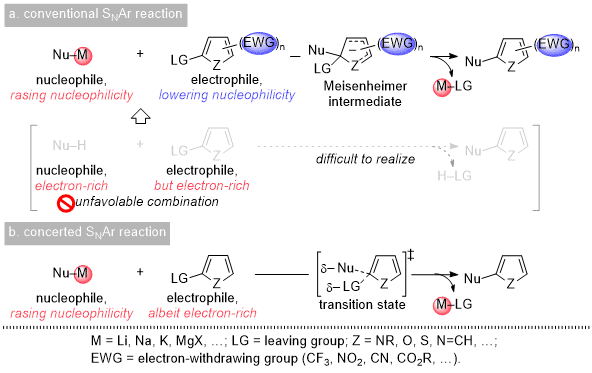
Scheme 1. Conventional and concerted SNAr reactions
Our research group has been engaged in developing new Lewis-acid-catalyzed reactions, of which indium Lewis acids serve as the genesis of our study.
14,15 In 2000, we reported for the first time that indium salts are suited for activating the C≡C bond of alkynes;
16 the inspiration of our indium chemistry stems from the unique carbophilic nature of allylindium reagents, which can survive under aqueous conditions without undergoing hydrolysis and thus cleavage of the C-In bond, and can successfully add to carbonyl compounds.
17 Since our initial report, we have been continuing the use of indium salts as Π-Lewis acid catalysts for the activation of C≡C and C=C bonds,
18 and the resulting indium-activated carbon electrophiles have been mainly utilized for the S
EAr (
E = electrophilic) reaction using (hetero)aryl nucleophiles.
14f,19 Even a C-C bond, albeit requiring the assistance of a directly connecting heteroaryl ring, can be cleaved by indium salts.
19b,e,f,g,h,20 A series of these studies are based on our research project: "
Activation of Hydrocarbon Functional Groups Classified into C≡C, C=C, C-C, and C-H21 mainly by Indium Lewis Acids".
22The C-C bond cleavage, described above, is observed during the indium-catalyzed three-component alkylation of pyrroles or indoles with alkynes or carbonyl compounds and nucleophiles (Nu) (Scheme 2). We considered at the time that the coordination of the heteroaryl ring to the indium salt (InX3 = In) would be crucial to trigger the C-C bond cleavage. Furthermore, it was anticipated that the coordination should occur on the π-face rather than the heteroatom of the heteroaryl ring, due to the carbophilicity of In. We therefore envisioned that utilizing this coordination mode could enable the direct activation of the heteroaryl ring itself. Some findings obtained by investigations performed based on the working hypothesis are discussed and summarized in the ensuing sections.
Scheme 2. C-C Bond cleavage triggered by the π-face coordination of the heteroaryl ring to the indium salt
Heteroaryl-Heteroaryl Bond-Forming Reaction
The first achievement is the S
NAr-based heteroaryl-heteroaryl bond-forming reaction
23 presented in the original article.
24 The topics that have not been discussed in the original article and that are crucial for this Discussion Addendum are addressed here. Interestingly, only the electron-donating OMe group serves as a leaving group (Scheme 3; Ac = acetyl), in marked contrast to the typical S
NAr reaction where EWGs like Cl and NO
2 act as leaving groups. With the more electron-rich 2,5-dimethoxythiophene (
2b), the reaction occurs even at room temperature (rt).
Scheme 3. Effect of leaving groups
Compounds 2 are electrophiles that react with electron-rich 1a. However, 2 is clearly more favorable with higher Π-electron density. The behavior of 2 might at first seem unusual but provides a useful insight into a reaction mechanism. The result of Scheme 4, giving 3ba-d and 3'ba-d from deuterated 1,2-dimethylindole (1b-d), is also crucial for mechanistic considerations.
Scheme 4. Indium-catalyzed SNAr reaction of 2-methoxythiophene with 1,2-dimethylindole-d
Proposed reaction mechanisms that take the above observations into account are shown in Scheme 5 by the reaction of HetAr-D
1-
d with
2a. First up is the Π-face coordination of
2a to
In to give complex
4a, in which
In serves as a transient EWG to make
2a electrophilic enough and thus to induce the nucleophilic attack of
1-
d via path a and/or b, giving allylindium-type intermediates
5-
d and/or
5'-
d, respectively. Subsequent D
+ transfer to their α and/or γ sites to give
6-
d and
6'-
d25 followed by the aromatizing elimination of MeOH(D) yields
3-
d and
3'-
d. This reaction mechanism nicely explains the results of Schemes 3 and 4. Thus, the role of the MeO group is to enhance the π-electron density of the thiophene ring and facilitate the complexation of
2a with electrophilic
In. The 23% loss of the D atom observed should be attributed to the final step that can release both MeOH and MeOD. Moreover, the formation of
3-
d and
3'-
d due to the proposed deuteration of the C-
In bond supports the probability of π-face coordination mode
4a in which the carbon atoms of
2a directly interact with
In.

Scheme 5. Proposed reaction mechanisms
Worthy of note is that neither heteroaryl-metal nucleophiles nor EWGs-substituted heteroaryl electrophiles are necessary for this strategy. Moreover, the S
NAr reaction between two electron-rich heteroarenes is unique.
26 To the best of our knowledge, no catalytic S
NAr reaction involving heteroarene-metal π-complexs
27 has been presented other than reports based on our strategy (
vide infra).
26 Next, we envisioned that electron-rich compounds other than
1 could be suitable for the indium-catalyzed S
NAr reaction.
Nitrogen-, Oxygen-, and Sulfur-Heteroaryl Bond-Forming Reactions
The electron-rich compound that we next focused on is an amine, thereby allowing the synthesis of a broad range of heteroarylamines.
28 Representative results obtained when using MeO-(benzo)thiophenes
2 are presented in Table 1. In(NTf
2)
3 is more effective than In(OTf)
3 for these reactions. As nucleophiles
7, primary and secondary alkyl/aryl amines with cyclic/acyclic structures can be used. With 3-bromo-4-methoxythiophene (
2d), the MeO-selective amination uniquely occurs, thus leaving the Br group intact in product
8gd. If low-boiling amines are desired as nucleophiles, their salts,
7m and
7n, are good choices (
8me and
8ne). This reaction features high functional group compatibility: besides functional groups listed in Table 1, C(
sp2)-I, -CF
3, -CN, -OH, C(
sp3)-OH, pyridyl, thiazolyl, benzyl, and C=C are all tolerated.
Table 1. Indium-catalyzed SNAr amination of MeO-(benzo)thiophenes
Heteroaryl electrophiles 2 besides MeO-(benzo)thiophenes are also capable of participating in the reaction (Table 2).
Table 2. Indium-catalyzed SNAr amination with (benzo)furyl-, pyrrolyl-, and indolyl-based electrophiles
Furthermore, alcohols and thiols can be used instead of amines in this strategy,
29 and Scheme 6 displays representative examples.
Scheme 6. Indium-catalyzed SNAr alkoxylation and thiolation
Nitrogen-Heteroaryl Bond-Forming Reaction Followed by Carbon-Heteroaryl Bond-Forming Annulation
We expected that combining two of our indium-catalyzed reactions, one of which is the S
NAr amination
28 and the other is the addition of heteroarenes to a C≡C bond,
19 could provide expedient access to heteroaryl[
b]quinolines (HA[
b]Qs).
30 A working hypothesis is illustrated in Scheme 7. The initial step would be the S
NAr amination of
2 by
13a via Π-coordination
4 to afford
14. The C≡C bond of
14 would then be activated as in
15 to induce the intramolecular S
EAr addition of the heteroaryl ring, thereby providing
16. Aromatization of
16 would result in the generation of HA[
b]Qs
17.
Scheme 7. A working hypothesis for constructing HA[b]Qs
To verify the working hypothesis, the annulation of o-ethynylaniline (13a) with 3-methoxybenzothiophene (2e) shown in Scheme 8 was tested.
Scheme 8. Indium-catalyzed annulation of o-ethynylaniline or o-acetylaniline with 3-methoxybenzothiophene
The treatment of
13a and
2e with 5 mol% of In(NTf
2)
3 under the heating conditions delivered the desired benzothieno[3,2-
b]quinoline
17ae, albeit in a low yield of 11%. Switching the catalyst to In(ONf)
3 (Nf = SO
2C
4F
9) gave not only
17ae but also a small amount of
o-acetylaniline (
18a). The carbonyl group in
18a was assumed to be formed by indium-catalyzed hydration of the C≡C bond with H
2O present in the reaction mixture. Hence, it was proposed that
17ae could be formed via the S
NAr amination of
2e with
18a followed by intramolecular nucleophilic addition of the benzothienyl ring to the carbonyl group and dehydration. Based on this proposal, the reaction of
13a with
2e was carried out with added H
2O, and as anticipated, the yields of both
17ae and
18a were raised. Prolonging the reaction time from 24 h to 36 h further improved the yield of
17ae to 61% with the complete consumption of
18a. Due to these results, we conducted the direct annulation of
18a with
2e and obtained
17ae in 92% yield by using catalyst InBr
3, as also shown in Scheme 8. These results show that InX
3 activates the benzothienyl ring of
2e and the carbonyl group of
18a. The ability for both the Π- and σ-electron-welcoming characteristics of InX
3 presents diverse opportunities for reactions.
18b We have utilized this reactivity
19c,d,i,j,20,31 and further demonstrate indium's utility as a two-way activator. Representative results mainly focusing on the scope of
18 are thus collected in Table 3. For example, 2-propyl (
18b), CF
3 (
18c), and a series of aryl (
18d-
h) groups are available as R
1. The carbonyl group between two aryl rings (
18h), the OH group (
18i), and the acetal moiety (
18j) remained untouched. Various thieno[2,3-
b]quinolines
17ka-
ga can be also obtained from
2a instead of
2e.
32Table 3. Indium-catalyzed synthesis of HA[b]Qs
This method followed by a two-step transformation enables to synthesize cryptolepine derivatives, which represent a significant structural motif with anti-malarial and -cancer activities.
33 Thus, for instance, the indium-catalyzed annulation of
18c with
2j can be used to construct
17cj, which, when followed by the methylation and treatment with aq. Na
2CO
3, delivers
20 (Scheme 9).
Scheme 9. Application to synthesis of a cryptolepine derivative
Formal N-Arylation and N-Alkylation of Pyrroles
The chemistry of the indium-heteroarene π-complex can be further applied to a distinct type of reaction: indium-catalyzed formal N-arylation and N-alkylation of pyrroles.
34 This transformation involves a unique nitrogen-nitrogen exchange strategy, or in other words, a pyrrole-ring opening-closing strategy. A working hypothesis that was developed before embarking on this study is depicted in Scheme 10.
Scheme 10. A working hypothesis for formal N-arylation and N-alkylation of pyrroles
Upon the treatment of pyrrole (
21a) and amine
7 with an indium catalyst (
In), we expected the sequence of reaction steps illustrated in Scheme 10. Thus, coordination of
21a to
In would make
21a electrophilic and promote reaction with
7, giving the enamine intermediate
24 via
23. Isomerization of
24 to imine
25 and coordination of its nitrogen atom to
In would generate
26, which could participate in ring opening and closing to produce
28 that incorporates the nitrogen atom of
7. This sequence can be regarded as a variation on the Paal-Knorr pyrrole synthesis.
35 The bond formation when preparing
N-aryl- and
N-alkylpyrroles from pyrroles is usually made directly on the nitrogen atom. Accordingly, this indium-catalyzed process is totally distinct from the general approach and thus unique.
36The N-arylation and N-alkylation of pyrroles are carried out by two methods: method A with solvent 1,4-dioxane and method B with no solvent. Representative results are summarized in Table 4.
Table 4. Indium-catalyzed formal N-arylation and N-alkylation of pyrroles
The synthesis of 28ao-go indicates the scope of pyrroles 21 that can be formed in the reaction, and the other products found in Table 4 demonstrate the scope of amines 7. When 1,2-phenylenediamine (7q) is used, only one amino group reacted with 2-methylpyrrole (21b) to yield 28bq. With 5-amino-2-methylindole (7t), the N-arylation chemoselectively occurred on the pyrrole ring, and the indolyl N-H thus remained unmodified, producing 28ct in a high yield. No racemization was observed in the reaction of (S)-1-phenylethylamine (7w), suggesting that no pyrrolyl-N-C bond-forming step is involved in this reaction.
Although the results of mechanistic studies are not provided herein, it was demonstrated that the mechanistic proposal of Scheme 10 is plausible.
34Closing Remarks
This Discussion Addendum started with a brief history of the indium π-Lewis acid that is crucial in promoting our original chemistry and influencing a subsequent series of studies utilizing the indium-heteroarene π-complex (Figure 1). Since the first discovery of the heteroaryl-heteroaryl bond-forming reaction in which the π-complex between
In and the MeO-substituted heteroarene participates, we have developed a number of new reactions: the nitrogen-, oxygen-, and sulfur-heteroaryl bond-forming reactions as well as the annulation reaction through the nitrogen-heteroaryl bond formation followed by the intramolecular carbon-heteroaryl bond formation. These reactions are unique because of occurring catalytically on electron-rich heteroaryl rings and should thus be classified as a distinct type of S
NAr reaction from the conventional and concerted ones.
37 Moreover, the π-complex has been demonstrated to be applicable to the formal N-arylation and N-alkylation of pyrroles.
Figure 1. Indium-heteroarene π-complex
We are continuing to dedicate our efforts to the chemistry of the indium-heteroarene π-complex, with the anticipation of presenting our new findings in upcoming articles.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved