Org. Synth. 2023, 100, 418-445
DOI: 10.15227/orgsyn.100.0418
3-Component Approach to 1,5-Dihydro-2H-pyrrol-2-one Heterocycles
Submitted by Nicholas P. Massaro and Joshua G. Pierce*
1Checked by Colin Knus, Jorge A. González, and Cristina Nevado
1. Procedure (Note 1)
A. Methyl (Z)-2-hydroxy-3-phenylacrylate (1). A 250 mL round-bottomed flask (24/40 joint) with a 35 mm egg -shaped Teflon-coated stir bar is charged with phenylpyruvic acid (2.50 g, 15.2 mmol, 1.0 equiv) (Note 2). The flask is sealed with a rubber septum and an argon balloon is connected through a needle and used to purge the atmosphere with argon. Anhydrous DMF (76 mL) (Note 3) is added via syringe to the flask (Figure 1a). The flask is placed in an ice-water bath at 0 ℃, and the solution is stirred at 500 rpm. Then DBU (2.3 mL, 15.2 mmol, 1.0 equiv) (Note 4) is added via syringe in one portion over 30 sec followed by addition of iodomethane (4.8 mL, 76.2 mmol, 5.0 equiv) (Note 5) also in one portion over 30 sec, after which point the transparent solution becomes yellow (Figure 1b).
Figure 1. a) Solution of phenylpyruvic acid in DMF at 0 ℃; b) After addition of DBU and iodomethane (photos provided by submitters)
Product formation is observed by TLC analysis (Figure 2) on silica gel using with EtOAc-hexanes (2:3; v/v) (Note 6) as eluent and visualizing with both UV and ceric ammonium molybdate (CAM) stain (Figure 2b) (Note 7). After stirring for 4 h, the reaction is cooled to 0 ℃ and quenched by the addition of an aqueous solution of HCl (80 mL, 1 M, over one min) (Figure 2c) (Note 8). Diethyl ether (80 mL) (Note 9) is added, and the mixture is allowed to warm to room temperature (Figure 3a).
Figure 2. a) TLC analysis under UV light. Lane 1 is phenylpyruvic acid, lane 2 is the co-spot and lane 3 is the reaction mixture; b) TLC analysis with ceric ammonium molybdate (CAM) staining; c) Quenched reaction after addition of 1 M (photos provided by submitters)
Figure 3. a) Quenched reaction at room temperature with HCl (80 mL, 1 M) and diluted with diethyl ether (80 mL). b) Quenched reaction transferred to separatory funnel and visible two layers. c) Filtration of organic layer over sodium sulfate into a 500 mL round-bottom flask. d) Concentration of crude filtrate onto 10 g of silica gel on rotary evaporator (photos provided by submitters)
The contents are transferred to a 250 mL separatory funnel (Figure 3b), and the organic layer is separated and filtered through 10 g of sodium sulfate (Note 10) that is placed in a glass funnel plugged with a small amount of cotton into a 500 mL round-bottomed flask (24/40 joint). The crude reaction mixture is extracted with diethyl ether (3 x 80 mL), and the combined ether layers are filtered over sodium sulfate into the 500 mL round-bottomed flask (24/40 joint) (Figure 3c). Silica gel (10 g) is added to the flask, and the mixture is concentrated in vacuo (40 ℃, 356 to 23 mmHg) (Figure 3d) to provide a pale-yellow solid (Figure 4a), which is directly loaded onto a 90 g silica flash column (Note 11) (Figure 4b). Sand is carefully added to the top of the column, providing an equal height to that of the dry loaded silica (Figure 4c). The flash column is eluted with a gradient of hexanes and ethyl acetate (Note 12) , and the product eluted in fractions 32-65 as determined by TLC analysis on silica gel with 2:3 (EtOAc-hexane) (Note 7) (Figure 4d).

Figure 4. a) Crude filtrate concentrated onto 10 g of silica gel. b) addition of crude mixture on silica followed by sand to pre-equilibrated 90 g silica gel flash column with approximately 100 mL hexanes. c) Appearance of flash column after product had fully eluted. d) Fractions containing product are confirmed by TLC analysis on silica gel with EtOAc-hexane (2: 3 v/v) as eluent and visualizing with both UV and CAM staining. Product 1 is collected in fractions 32 to 65 (photos provided by submitters)
Fractions containing the product are transferred to a 1 L round-bottomed flask and concentrated in vacuo (40 ℃, 135 mmHg). This resultant oil is transferred to a pre-weighed 20 mL scintillation vial using ethyl acetate (7 mL). Ethyl acetate is then removed in vacuo (40 ℃, 98 mmHg) (Figure 5a) to provide compound 1 as a clear oil. A stream of argon is blown directly on the oil (Figure 5b), which provides a waxy white solid (Figure 5c). Residual solvent is removed by high vacuum (<3 mmHg) to generate 1.34 g (50%) of pure methyl (Z)-2-hydroxy-3-phenylacrylate (1) (Notes 13 and 14).
Figure 5. a) Concentration of 1 into a 20 mL scintillation vial. b) Solidification of 1 under an argon stream. c) Isolated 1 as a waxy solid (photos provided by submitters)
B. 1-(2,4-Dimethoxybenzyl)-3-hydroxy-5-pentyl-4-phenyl-1,5-dihydro-2H-pyrrol-2-one (2). A 100 mL round-bottomed flask (14/20 joint) with a 2.0 cm egg-shaped Teflon-coated magnetic stir bar is charged with methyl (Z)-2-hydroxy-3-phenylacrylate (1) (1.5 g, 8.42 mmol, 1.00 equiv), 200 mg of powdered activated 5 Å molecular sieves (Note 15) and anhydrous dichloromethane (17.0 mL) (Note 16). Hexanal (1.13 mL, 9.26 mmol, 1.1 equiv) (Note 17) is added to the solution via syringe over 30 sec. An addition funnel is equipped with a rubber septum and attached to the round-bottomed flask, and the system is purged with nitrogen and then placed under nitrogen atmosphere via a needle through the septum. The flask is placed in an ice-water cooling bath, and the addition funnel is charged with a solution of 2,4-dimethoxybenzylamine (1.42 mL, 9.26 mmol, 1.1 equiv) (Note 18) in anhydrous dichloromethane (7 mL). The reaction mixture is stirred (450 rpm) and the 2,4-dimethoxybenzylamine is added dropwise over a period of 25 min (Figure 6a). The solution slowly turned from pale yellow (Figure 6b) to a darker yellow (Figure 6b). The reaction is monitored by TLC using EtOAc:hexanes (2:3) as the mobile phase, with visualization by UV light (Figure 6c) or by CAM staining with heating (Figure 6d).
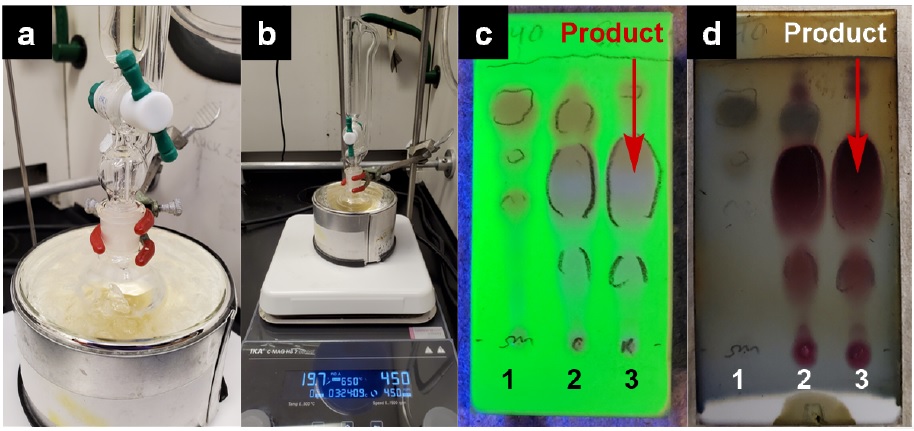
Figure 6. a) Initial setup of the reaction. b) After 25-minute slow addition of 2,4-dimethoxybenzylamine with addition funnel. c) TLC UV visualization 1 hour after addition of 2,4-dimethoxybenzylamine. Lane 1 contains starting material (1), lane 2 is the co-spot, and lane 3 is the reaction mixture. d) TLC CAM staining of TLC shown in (c). Product (2) Rf = 0.60 using EtOAc:hexanes (2:3 v/v) as eluent (photos provided by submitters)
After 3 h of stirring at 0 ℃ the crude reaction is filtered through a 50 mL glass fritted filter into a 100 mL round-bottomed flask (14/20) to remove the powdered molecular sieves (Figure 7a), and the filtered sieves are rinsed with additional dichloromethane (15 mL). The filtrate is concentrated in vacuo at 30 ℃ (11-23 mmHg) to provide a grayish/yellow oil. Residual solvent is removed by high vacuum (3 mmHg), which results in formation of an amorphous solid (Figure 7b). The flask is removed from high vacuum, and a mixture of dichloro-methane : hexanes (50 mL, 1:9 v/v) is added, which does not dissolve the crude residue (Figure 7c).
Figure 7. a) Filtration of crude reaction to remove 5 Å molecular sieves; b) Crude residue after removal of solvent in vacuo and high vacuum; c) Crude mixture after addition of 50 mL of dichloromethane:hexanes (1:9 v/v) mixture (photos provided by submitters)
This mixture is sonicated for 5 min at 25 ℃ to dissolve the impurities, and the solution became turbid white due to the desired product being insoluble in the solution (Figure 8a). The white precipitate is isolated by vacuum filtration through a 50 mL (14/20) joint glass fritted filter (Figure 8b). Product 2 is transferred via spatula to a 20 mL scintillation vial that is subsequently placed under high vacuum (<3 mmHg) until a constant mass is obtained. The isolated product (2) is obtained as a white claylike solid (2.80 g, 84%) (Figure 8c) (Notes 19 and 20).
Figure 8. a) Sonication of crude reaction mixture in 100 mL round-bottom flask containing dichloromethane:hexanes mixture (1:9 v/v). b) Filtration of white solid precipitate obtained from (a). c) Isolated desired compound as a pure white claylike solid (photos provided by submitters)
C. 3-Hydroxy-5-pentyl-4-phenyl-1,5-dihydro-2H-pyrrol-2-one (3). To a 24/40 joint round-bottomed flask containing 2 (5.00 g, 12.6 mmol, 1.0 equiv) with a 4.0 cm egg-shaped stir bar is added anhydrous dichloromethane (100 mL). The flask is equipped with a 50 mL addition funnel and a rubber septum. The system is purged with nitrogen through a needle through the septum and then a nitrogen atmosphere is maintained. Trifluoroacetic acid (25 mL) (Note 21) is added to the addition funnel through the septum using a glass syringe over 30 sec. The flask is placed in an ice-water cooling bath (Figure 9a), and the trifluoroacetic acid is added dropwise over 45 min (Figure 9b). During addition of the trifluoroacetic acid, the solution slowly turns from pale yellow to a pale orange. Upon completion of the addition, the reaction is stirred for 1 h while allowing the cooling bath to reach 10 ℃ as the ice melts (Figure 9c). The cooling bath is removed, and the reaction is allowed to stir for another 5 h, after which time the reaction mixture is a deep purple color (Figure 9d).

Figure 9. a) Beginning of reaction with the start of slow addition of trifluoroacetic acid over 45 min. b) Completion of addition of trifluoroacetic acid over 45 min at 0 ℃. c) Reaction after stirring for 1 h to 10 ℃ when ice-water bath is removed. d) Reaction after stirring at room temperature for 5 h (photos provided by submitters)
The progression of the reaction is monitored by TLC analysis on silica gel using EtOAc-hexane (1:1 v/v) as eluent and visualizing with both UV and CAM staining (Note 22). After a total reaction time of 7 h, the reaction is removed from the stirring plate. The stir bar is removed and rinsed with dichloromethane (5 mL). The crude reaction mixture is concentrated in vacuo (Figure 10a), (40 ℃, gradually lowering to 11-23 mmHg). The crude residue is then placed under high vacuum (<3 mmHg) for 2 h) to remove residual solvent (Figure 10b). A mixture of dichloromethane:hexanes (200 mL, 1:9 v/v) is added to the flask. The resulting mixture is briefly sonicated (5 min), after which time, hexanes (50 mL) is added to the mixture. The resulting mixture is once again briefly sonicated (5 min). The flask is equipped with a rubber septum and stored at -20 ℃ for 2 h to allow the pale purple precipitate to settle on the bottom of the flask (Figure 10c). The solid is removed by vacuum filtration with a 150 mL (24/40) glass fritted filter (Figure 11a), which provides a pale purple solid impurity (Figure 11b).

Figure 10. a) Solvent and trifluoroacetic acid are removed by rotary evaporation and high vacuum. b) Crude solid obtained after rotary evaporation. c) Crude mixture after addition of 200 mL of dichloromethane:hexanes (1:9 v/v) with brief sonication followed by addition of another 50 mL hexanes and brief sonication providing a purple precipitate (photos provided by submitters)
Figure 11. a) Removal of purple solid impurity by vacuum filtration. b) Isolated purple solid impurity from trituration with dichloromethane:hexanes and sonication (photos provided by submitters)
The filtrate containing the desired product is collected in a 24/40 joint 500 mL round-bottomed flask. The desired product begins to precipitate immediately (Figure 12a) (Note 23). This new white precipitate is then collected by vacuum filtration with another 150 mL 24/40 joint fritted filter providing a pasty white solid (Figure 12b). The resulting filtrate begins to form a precipitate of the desired product as a gelatinous solid (Figure 12c). This solid is collected by filtration once again using the same fritted filter as used to collect the desired product earlier (Figure 12d). The collected white solid from the two filtrations is left in the filter, washed with hexanes, and dried under vacuum in the filter (Figure 12e). The filtrate flask is removed, and a clean 24/40 joint 500 mL round-bottomed flask is attached to the 150 mL fritted glass filter. The solid in the filter is dissolved in acetone (Figure 12f) (Note 24) and pulled into the 500 mL round-bottom flask by vacuum (Figure 12g). Concentration in vacuo provides the crude product as a solid (Figure 12h), (40 ℃, gradually lowering to full vacuum of approximately 11-23 mmHg).

Figure 12. a) Filtrate obtained after removal of purple solid impurity. b) Filtration of white solid precipitate generated in filtrate seen in (a). c) Gelatinous solid obtained from filtrate performed in (b). d) Subsequent filtration of precipitate seen in (c). e) Combined white solid obtained from filtration and hexane washes. f) White pasty solid is eluted with reagent grade acetone by vacuum. g) Filtrate obtained from dissolving desired compound in acetone and collecting in flask. h) Removal of acetone by concentration with rotary evaporation (photos provided by submitters)
The flask containing the product is placed under high vacuum (<3 mmHg) (Figure 13a) to remove residual solvent and provide a pale white, pasty solid (Figure 13b). The product is transferred via spatula to a pre-weighed 20 mL scintillation vial, which is subsequently placed under high vacuum (<3 mmHg) to provide 1.96 g (63%) of 3-hydroxy-5-pentyl-4-phenyl-1,5-dihydro-2H-pyrrol-2-one (3) (Notes 25 and 26).
Figure 13. a) Removal of residual solvent by high vacuum. b) Product 3 obtained as a pure white solid (photos provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out regarding each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
phenylpyruvic acid,
1,8-diazabicyclo[5.4.0]undec-7-ene,
iodomethane,
molecular sieves 5Å,
hexanal,
2,4-dimethoxybenzylamine,
trifluoroacetic acid,
sodium sulfate,
dichloromethane,
hexanes,
ethyl acetate,
acetone,
diethyl ether, 1M
hydrochloric acid solution, and silica gel.
2.
Phenylpyruvic acid, >90.0% was purchased from TCI and used as received.
3.
Dimethylformamide (
DMF) was purchased from Fisher (HPLC grade >99.5%), dried by mBraun SPS, and stored under argon.
4.
1,8-Diazabicyclo[5.4.0]undec-7-ene (99%) was purchased from Thermo Scientific and used as received.
5.
Iodomethane (99%) was purchased from Thermo Scientific and used as received.
6.
Hexane (technically pure) was purchased from Thommen-Furler.
Ethyl acetate (technically pure) was purchased from Thommen-Furler.
7. The
phenylpyruvic acid does not move from the baseline under these TLC conditions. The product possesses an R
f = 0.71 (highly UV active spot and ceric
ammonium molybdate (
CAM) active spot).
8.
HCl solution was prepared from a solution (37%) purchased from Merck.
9.
Diethyl ether (>99.8%) was purchased from Honeywell and used as received.
10.
Sodium sulfate anhydrous (>99.0%) was purchased from Merck.
11. Silica gel 60 M (0.04-0.063 mm) was purchased from Macherey Nagel. Please note that bumping can easily occur during preparation of the compound-infused silica. Considerable product degradation was detected during the preparation of the compound-infused silica. Approximately 100 mL of
hexanes was used to complete the loading of the compound-infused silica onto the column. The chromatography column, prior to the addition of the compound-infused silica, was 10 cm in length. The column diameter was approximately 5.5 cm.
12. The flash column is eluted with a step gradient of 100 mL
hexanes, 200 mL 2.5%
EtOAc:
hexanes, 200 mL 5.0%
EtOAc:
hexanes, 200 mL 7.5%
EtOAc:
hexanes and 600 mL of 10%
EtOAc:
hexanes (
Note 12).
Methyl (Z)-2-hydroxy-3-phenylacrylate (
1) began appearing in TLC fractions when 10%
EtOAc:
hexanes was the mobile phase, so this mixture was maintained until all compound eluted from the flash column. Fractions of 25 mL fractions were collected in 18 x 150 mm glass test tubes. The product appeared in fractions 32-65 as observed by TLC analysis on silica gel with 2:3
EtOAc-
hexane as eluent and visualizing with both UV and CAM staining.
13.
Methyl (Z)-2-hydroxy-3-phenylacrylate (
1): mp 56-62 ℃ (provided by submitter). Compound (
1) exists 85% as the enol tautomer and 15% as the keto tautomer. Enol tautomer:
1H NMR
pdf (500 MHz, CDCl
3) δ: 7.80 (dt,
J = 8.2, 1.4 Hz, 2H), 7.40 (t,
J = 7.7 Hz, 2H), 7.32 (td,
J = 7.4, 1.3 Hz, 1H), 6.58 (s, 1H), 6.54 (s, 1H), 3.95 (d,
J = 1.4 Hz, 3H); Keto tautomer:
1H NMR
pdf (500 MHz, CDCl
3) δ: 7.39-7.36 (m, 2H), 7.27 (t,
J = 7.2 Hz, 2H), 4.16 (s, 2H), 3.87 (d,
J = 1.2 Hz, 3H); Enol tautomer:
13C NMR
pdf (125 MHz, CDCl
3) δ: 166.9, 139.2, 134.2, 130.0 (2C), 128.6 (2C), 128.1, 111.3, 53.4; Keto tautomer:
13C NMR
pdf (125 MHz, CDCl
3) δ: 191.1, 161.5, 131.7, 130.5, 129.9 (2C), 128.9 (2C), 127.6, 53.2, 45.8. IR (film) ν
max: 3381, 3053, 2057, 1256, 1573, 1494, 1450, 1440, 1390, 1344, 1330, 1243, 1199, 1184, 1114, 1032,980, 918, 882, 867, 840, 775, 748, 689, 635, 595, 554, 481, 459, 417, 404 cm
-1. HRMS-ESI-[M+H]
+ calculated for [C
10H
11O
3]
+: 179.0703, found: 179.0702.The purity of (
1) was determined to be 98 wt% by qNMR
pdf using
ethylene carbonate (Sigma Aldrich, standard for quantitative NMR, TraceCERT) as the internal standard.
14. When the reaction was performed on a larger scale, a yield of 2.48 g (92%) was isolated with 88% purity.
15. Molecular sieves 5 Å, 8 to 12 mesh were purchased from Thermo Scientific. Sieves were grinded, and the powder was activated at 200 ℃ for 14 - 16 h. The powder was used after fresh activation.
16. Anhydrous
dichloromethane was purchased from Honeywell (HPLC grade >99.5%), dried by mBraun SPS, and stored under argon.
17.
Hexanal (98%) was purchased from Thermo Scientific and used as received.
18.
2,4-Dimethoxybenzylamine (98%) was purchased from Thermo Scientific and used as received.
19.
1-(2,4-Dimethoxybenzyl)-3-hydroxy-5-pentyl-4-phenyl-1,5-dihydro-2H-pyrrol-2-one (
2): mp 145-146 ℃.
1H NMR
pdf (500 MHz, CDCl
3) δ: 7.57 (d,
J = 7.7 Hz, 2H), 7.36 (t,
J = 7.8 Hz, 2H), 7.25 (t,
J = 7.6 Hz, 1H), 7.18c (d,
J = 8.2 Hz, 1H), 6.95 (s, 1H), 6.48-6.43 (m, 2H), 5.01 (d,
J = 15.0 Hz, 1H), 4.37 (t,
J = 3.7 Hz, 1H), 4.29 (d,
J = 15.0 Hz, 1H), 3.84 (s, 3H), 3.80 (d,
J = 0.9 Hz, 3H), 1.95 (ddt,
J = 15.5, 11.7, 4.3 Hz, 1H), 1.79-1.69 (m, 1H), 1.13 (h,
J = 7.3 Hz, 2H), 1.09-1.00 (m, 2H), 0.93-0.81 (m, 2H), 0.75 (t,
J = 7.25 Hz, 3H).
13C NMR
pdf (125 MHz, CDCl
3) δ: 167.5, 160.7, 158.5, 142.1. 132.0, 131.0, 128.6 (2C), 127.4 (2C), 127.4, 121.2, 117.6, 104.5, 98.6, 56.9, 55.5, 38.4, 31.7, 28.6, 22.6, 20.8, 14.1. IR (film) ν
max: 3171, 2918, 1651, 1610, 1508, 1444, 1383, 1336, 1308, 1261, 1235, 1208, 1166, 1126, 1109, 1031, 938, 855, 820, 792, 766, 729, 692, 652, 636, 555, 497, 464, 438, 423 cm
-1. HRMS-ESI-[M+H]
+ calculated for [C
24H
30NO
4]
+: 396.2169, found: 396.2168. The purity of (
2) was determined to be >99 wt% by qNMR
pdf using
ethylene carbonate (Sigma Aldrich) as the external standard.
20. A second reaction at full scale provided 2.59 g (78%) of the product with >97% purity.
21.
Trifluoroacetic acid (98.5%) was purchased from Honeywell and used as received.
22. UV visualization of the TLC plate proved to be inadequate for comparison of starting material consumption and product formation (Figure 14a). Initial TLC analysis performed immediately after addition of
trifluoroacetic acid showed significant starting material present on TLC (Figure 14b). The starting material appears purple with CAM staining and heating. As the reaction progresses and the Dmb group is cleaved, the appearance of the 'charred' product spot differs significantly from the 'charred' starting material spot, which can be used to assess completion of the reaction (Figure 14c).
Figure 14. a) TLC UV visualization immediately after slow addition of trifluoroacetic acid. b) CAM stain visualization with charring immediately after slow addition of trifluoroacetic acid. c) TLC CAM visualization with charring after 5 h of stirring at room temperature. Product (2) Rf = 0.41 using EtOAc:hexanes (1:1 v/v) as eluent (photos provided by submitters)
23. In rare cases, the submitters observed that immediate precipitation did not occur after removal of the purple solid impurity from the first filtration as shown in Figure 11, which is suspected to be due to residual
trifluoroacetic acid. In such cases, precipitation can be induced by concentrating the filtrate using a rotary evaporator providing a white solid with some pink/purple discoloration. The impure product can be purified by adding 200 mL of
dichloromethane:
hexanes (1:9; v/v), briefly sonicating, adding another 50 mL
hexanes, and briefly sonicating once again. Concentration to a volume of approximately 100 mL provides the product as a white solid with less pink/purple discoloration. A sample of this filtered solid was taken for
1H NMR analysis confirming relatively high purity of product, which is also provided in this protocol. Finally, the collected product can be redissolved in a mixture of 200 mL
hexanes and 50 mL
dichloromethane. Upon sonication, the product forms as a white precipitate that can be isolated by vacuum filtration providing
3-hydroxy-5-pentyl-4-phenyl-1,5-dihydro-2H-pyrrol-2-one (
3) in similar yield.
24.
Acetone (>99.5%) was purchased from Merck and used as received.
25.
3-Hydroxy-5-pentyl-4-phenyl-1,5-dihydro-2H-pyrrol-2-one (
3): mp 168-169 ℃.
1H NMR
pdf (500 MHz, DMSO-
d6) δ: 9.85 (s, 1H), 8.58 (s, 1H), 7.59 (dd,
J = 8.1, 1.2 Hz, 2H), 7.38 (t,
J = 8.0 Hz, 2H), 7.24 (t,
J = 7.4, Hz, 1H), 4.48 (dd,
J = 7.4, 2.8 Hz, 1H), 1.79-1.66 (m, 1H), 1.33-1.07 (m, 7H), 0.79 (t,
J = 6.8 Hz, 3H).
13C NMR
pdf (125 MHz, DMSO-
d6) δ: 168.2, 143.5, 132.5, 128.4 (2C), 126.9 (2C), 126.8, 122.0, 52.8, 33.1, 31.1, 23.4, 22.0, 13.8. IR (film) ν
max 3260, 2997, 2955, 2856, 1698, 1671, 1497, 1429, 1406, 1360, 1296, 1226, 1103, 1063, 1028, 895, 870, 767, 696, 640, 477, 439, 425 cm
-1. HRMS-ESI-[M+H]
+ calculated for [C
15H
20NO]
+: 246.1489, found: 246.1490. The purity of (
3) was determined to be >99 wt% by qNMR
pdf using
ethylene carbonate (Sigma Aldrich) as the external standard.
26. A second reaction performed at half-scale provided 0.73 g (47%) of the product with 96% purity.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Nitrogen-containing heterocycles (
N-heterocycles) are a commonality among bioactive natural products often represented in alkaloids and other privileged scaffolds.
2,3 A frequently observed motif is a 5-membered
N-heterocycle, present in indoles, pyrroles, pyrrolidines and succinimides. In fact, 13 out of the 25 most frequent nitrogen heterocycles in U.S. FDA approved drugs are 5-membered.
4 The niche of 5-membered nitrogen containing heterocyclic natural products has supplied potent anticancer therapies such as vinblastine
5,6 as well as treacherous poisons such as strychnine.
7 Other examples exhibit higher oxidation levels of the pyrrolidine cores in the form of 2-pyrrolidone, represented by the highly toxic proteasome inhibitor salinospormide A
8 and angiogenesis inhibitor (-)-azaspirene.
9 Aside from metabolites within the environment, humans also possess numerous 5-membered nitrogen heterocycles such as bilirubin, a product of heme catabolism, which also can serve as a potent antioxidant (Figure 15).
10 Further, many examples highlight that natural products serve as direct sources of drugs or as inspiration in drug development.
11,12 In fact, within the class of 5-membered
N-heterocycles, there are numerous representations within the top 200 small molecule drugs ranking by sales.
13 Drugs such as Lipitor, sunitinib, and vonoprazan all represent pyrrole-based drugs. More oxidized succinimide and phthalimide cores are well represented as pharmaceuticals, with lurasidone, apremilast, pomalidomide, ethosuximide and lenalidomide as some key examples.
13,14
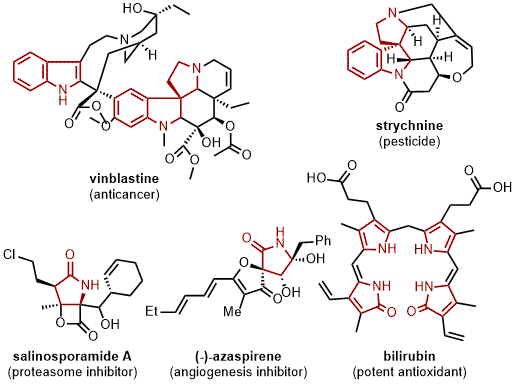
Figure 15. Representative 5-membered N-heterocyclic drugs and natural products
Figure 16. Marketed drugs that contain a 5-membered N-heterocycle with one nitrogen atom listed in the top 200 drugs sold in 2018 based on sales
In addition to the 5-membered
N-heterocycles that have made a mark in the pharmaceutical industry (Figure 16), there are others that represent the 2-pyrrolidone class specifically. Two key examples are leuprolide and goserelin, two hormone-based drugs to treat prostate and breast cancer that contain not only terminal 2-pyrrolidone, but various other 5-membered
N-heterocycles within their peptide sequences (Figure 17). In addition to the compiled list of top-200 drugs, there are other relevant bioactive compounds that contain the 2-pyrrolidone motif. Key examples include EBPC is a highly specific aldose reductase inhibitor,
15 holomcine,
16 doxapram,
17 lactacystin,
18 (
R)-rolipram,
19 and cotinine.
20,21 In addition to bioactivity, 2-pyrrolidone itself is a versatile building block while being a core motif in key commodity chemicals such as
N-methylpyrrolidone (NMP) and L-pyroglutaminol (Figure 18).
22
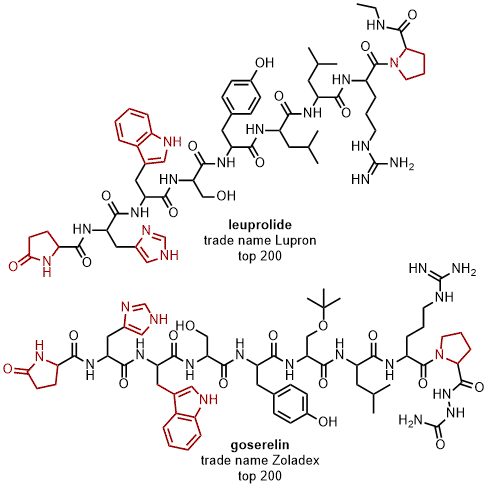
Figure 17. Representative examples of drugs containing a 2-pyrrolidone as well as other N-heterocycles showcased in the top 200 pharmaceutical products sold in 2018
Figure 18. 2-Pyrrolidone containing bioactive compounds and commodity chemicals
With regards to the overall impact of 5-membered
N-heterocycles, pyrrolidones have established themselves as a common motif for drug development ranging from hit-compounds to FDA approval. For this reason, efficient methodologies to access pyrrolidone cores is of high relevance. While numerous robust methods to prepare various 5-membered
N-heterocycles exist, such as the Huisgen reaction, Asinger reaction, and Groebke-Blackburn-Bienayme reaction, among others, there remains few examples of methodologies that access highly functionalized 2-pyrrolidones.
23,24,25 Of these approaches, many are linear strategies or multicomponent approaches that are often limited by their precursor preparation or the need for harsh reaction conditions. Multicomponent reactions provide a concise route to complex products from simple building blocks saving time and money by performing a reaction in one step over previously known multistep processes.
26,27
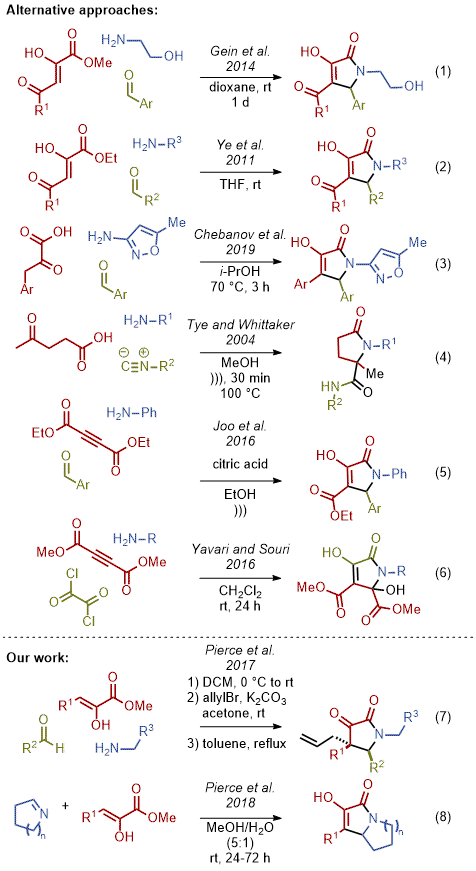
Figure 19. Multicomponent approaches to pyrrolidinone compounds
From the various multicomponent approaches to access highly functionalized heterocycles, including pyrrolidinones, the most common intermediate involves an imine.
28,29,30 Since the reactive imine is formed in situ, this strategy often provides a high level of modularity allowing for efficient synthesis of analogs for library generation.
31 Gein and co-workers have contributed significant efforts for the synthesis of these molecules via a multicomponent reaction that requires the need for an acyl electron withdrawing group with prolonged reaction time (Figure 19, equation 1).
32,33,34,35,36,37,38,39,40,41 Dekker and co-workers utilized this method to synthesize diverse pyrrolidinones as well.
42 Ye et al. showcased a very similar method also utilizing an acyl electron withdrawing group (Figure 19, equation 2).
43 In 2019, Chebanov and co-workers showcased a reaction directly with pyruvic acid instead of ester starting material, but the reaction requires near refluxing conditions in isopropanol (Figure 19, equation 3).
44,45,46 Other previous methods also incorporate a free carboxylic acid, such as Tye and Whittaker's strategy, although it requires refluxing temperatures with sonication (Figure 19, equation 4).
47 Joo and co-workers and Yavari and Souri independently synthesized similar pyrrolidinones utilizing symmetrical alkynes. Joo et al. utilized acidic conditions with sonication (Figure 19, equation 5)
48 while Yavari and Souri utilized a highly reactive oxalyl chloride (Figure 19, equation 6).
49Our research group established a method to access highly functionalized pyrrolidinones that provided the foundation for the synthesis of diverse quaternary pyrrolidine-2,3-diones (Figure 19, equation 7), bicyclic
N-heterocycles and natural products (Figure 19, equation 8).
50,51,52,53,54 The procedure described herein provides a representative example of this methodology. The methodology can be used to prepare products that lack an additional exocyclic carbonyl while progressing under extremely mild conditions.
In summary, N-heterocycles such as pyrrolidinones have shown to be of significant relevance in the pharmaceutical and fine chemical industry. Furthermore, their extensive presence in natural products makes it of high importance to develop scalable, efficient, and modular approaches to these cores. The procedure described herein provides an attractive means to synthesize highly functionalized pyrrolidinones.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Phenylpyruvic acid; (156-06-9)
DMF: N,N-Dimethylformamide; (68-12-2)
DBU: 1,8-Diazabicyclo[5.4.0]undec-7-ene; (6674-22-2)
Iodomethane; (74-88-4)
HCl: hydrochloric acid; (7647-01-0)
sodium sulfate; (7757-82-6)
EtOAc: ethyl acetate; (141-78-6)
Hexanes; (92112-69-1)
Methyl (Z)-2-hydroxy-3-phenylacrylate: Methyl 2‐oxo‐3‐phenylpropanoate; (1)(6362-58-9)
5 Å molecular sieves: Molecular sieves 5A, 8 to 12 mesh; (69912-79-4)
Hexanal; (66-25-1)
2,4-Dimethoxybenzylamine: 2,4-dimethoxybenzylamine; (20781-20-8)
Dichloromethane: methylene chloride; (75-09-2)
Trifluoroacetic acid: trifluoroacetic Acid; (76-05-1)

|
Joshua G. Pierce received a B.S. degree in Chemistry from the University of Pittsburgh in 2003, during which time he became involved in synthetic organic chemistry research in the lab of Professor Peter Wipf. Continuing at the University of Pittsburgh, he graduated with his Ph.D. in 2008 under the direction of Professor Wipf and then completed postdoctoral studies in the lab of Professor Dale L. Boger in 2009-2011. In 2012 he joined the Department of Chemistry at NC State University, where he is now the Howard J. Schaeffer Distinguished Professor of Chemistry, and Executive Director of the Integrative Sciences Initiative. His lab is focused on the synthesis and chemical biology of natural products. |

|
Nicholas P. Massaro received his B.A. and M.S. in Chemistry at the State University of New York at Oswego under the advisement of Professor Fehmi Damkaci, where he explored ligand modifications for the enhancement of Ullmann homocoupling reactions. He then moved to the University of Oklahoma, where he was awarded his Ph.D. in Chemistry in 2019 under the advisement of Dr. Indrajeet Sharma, focusing on diazo-derived carbenoid cascades for the synthesis of medium-sized rings. He then pursued a postdoctoral appointment with Dr. Joshua G. Pierce at NC State University focusing on the synthesis and testing of bioactive natural products and heterocycles. |

|
Colin Knus was born in Switzerland and joined the Nevado group in 2022 as a Lab Technician. He is currently a second year student of his Apprenticeship course. He enjoys Chemistry, travelling around on his motorbike, and snowboarding. |

|
Jorge A. González was born in Xalapa, Mexico. He completed his Undergraduate Degree in Chemistry at the National and Autonomous University of Mexico in 2011. He obtained his Ph.D. at the University of Edinburgh in 2016. He is currently a postdoctoral research associate in the group of Prof. Cristina Nevado. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved