Extending the Utility and Scope of the Reaction
Inspired by our chemoselective reduction protocol for converting amides to aldehydes,
2,3,4 sparing esters, and other functional groups prone to reduction, subsequent publications reported improved protocols for this transformation, including catalytic methods. Multiple groups extended the utility of the chemistry, including the formation of amines from acetamides, phenols from N,N-diethyl aryl O-carbamates, and the reductive cleavage reaction of heterocyclic carbamates. Many groups used the initially formed reduced intermediates to add amines, carbon nucleophiles, and phosphonates. The intermediates were also employed in Ugi and Mannich/Michael tandem reactions. Many of the reaction products are complex structures that are otherwise difficult to prepare.
In situ Generation of Cp2Zr(H)Cl
Zhao and Snieckus reported in 2014 a practical method for the
in situ generation of Cp
2Zr(H)Cl (Schwartz's reagent) from Cp
2ZrCl
2 and LiAlH(O
tBu)
3 to address the limitations associated with previously reported
in situ protocols and the limited shelf-life of the commercial reagent (Scheme 1).
5 In a comparative study against commercial Cp
2Zr(H)Cl they showed that the
in situ procedure proceeded with overall better reaction times and yields in the conversion of amides
1 to aldehydes
2. Representative examples
2a-
2m demonstrate again the excellent chemoselectivity of this reaction.
Scheme 1. Reduction of N,N-diethylamides to aldehydes using an in situ generated Schwartz reagent
Catalytic Amide Reductions with the Schwartz Reagent
To address the limited stability of the reagent, Donnelly et al. developed a catalytic version by using triethoxysilane ((EtO)
3SiH) as a mild stoichiometric reductant (Scheme 2).
6 This approach enables the efficient transformation of secondary amides
3 to imines
4 and tolerates a variety of functional groups. Under these conditions, 6-chloro-2-oxindole formed deoxygenated
4k after tautomerization of the initially formed imine. Mechanistic studies suggest that the turnover of the active [Zr]-H species is achieved through the metathesis of the Si-H and Zr-OR σ-bonds.
Scheme 2. Catalytic reduction of secondary amides with Schwartz's reagent
Scheme 3. Applications for the Cp2ZrCl2/DMMS catalytic system
In their 2023 study, Kehner et al. advanced another catalytic approach by employing the more stable Cp
2ZrCl
2 as a precursor for the zirconocene hydride catalyst, thereby circumventing the direct handling of the air- and moisture-sensitive Schwartz's reagent (Scheme 3).
7 Their method reduces secondary and tertiary amides using 5 mol% of Cp
2ZrCl
2 at room temperature. Dimethoxymethylsilane (DMMS) was utilized as a reductant in place of (EtO)
3SiH, to avoid the formation of the pyrophoric and toxic byproduct SiH
4. For secondary amides
5, this approach proved efficacious with aromatic amides, while the dialkyl amide N-benzylpentanamide gave a lower yield (41%). The chemistry was also applied to synthesizing indoles
8 from 2-indolinones
7. Notably, tertiary amides
9 could be reduced to imines
10 under these conditions through reductive transamination, adding an extra equivalent of a primary amine. In the case of aliphatic amide
12, the addition of secondary amines gave the corresponding enamines
13.
Conversion of α-Substituted Amides to Aldehydes with no or Minimal Erosion of Stereochemistry
In 2006, McGilvra et al. prepared β-hydroxy amides
14 employing a hydrogen bonding-catalyzed Mukaiyama aldol reaction.
8 The authors then used Cp
2Zr(H)Cl to convert amides
14 into aldehydes
15 with minimal erosion of the alpha stereocenter gained in the previous step. Key to the optimized conversion was the solvent change from THF to CH
2Cl
2 (Scheme 4), which significantly increased the reaction rate and prevented isomerization. The reduced diastereomeric ratio (dr) was attributed to the iminium intermediate, which can isomerize to an enamine when the reaction rate is slow.
Reductive Cleavage of Aryl O-Carbamates and Reductive Cleavage of Heterocyclic N-Carbamides
In 2013, Morin et al. reported a mild reductive cleavage method for conversion of aryl O-carbamates
16 to phenols
17 using Schwartz's reagent (Scheme 5).
9 Substituted phenol O-carbamates containing halogens, electron-withdrawing, and donating groups were studied. Reductive cleavages with the commercial and the in situ generated Cp
2Zr(H)Cl were equally efficient. The authors extended the scope of this reductive cleavage reaction to heterocyclic carbamates
18, which provided moderate to good yields for cleavage products
19 (Scheme 6).
Scheme 4. Conversion of amides to aldehydes with Cp2Zr(H)Cl with minimal erosion of stereochemistry
Scheme 5. Reductive cleavage of N,N-diethyl aryl O-carbamates to phenols using the in situ generated Schwartz reagent
Scheme 6. Reductive cleavage of heterocyclic carbamides using the Schwartz reagent
Chemoselective Conversion of Acetamides to Amines with Cp2Zr(H)Cl
While our original report focused on generating aldehydes from amides, the other species released in this reaction is the amine portion of the amide. Thus, Sultane et al. described in 2014 the selective N-deacetylation of acetamides
20 using the Schwartz reagent under mild conditions and with high chemoselectivity (Scheme 7).
10 This strategy demonstrated that N-deacetylation of aliphatic and heteroaromatic substrates is efficient and rapid, providing amines
21 in high yields from amide substrates with diverse electronic and steric properties. Epimerization was not observed during the synthesis of chiral amines
21e,
21f, and
21g.. Since Ts, Fmoc, Cbz, and Boc protection of amines was retained, this reductive procedure can be used in an orthogonal protection/deprotection strategy.
Scheme 7. Deacetylation of N-acetamides by the Schwartz reagent
Similarly, the application of Schwartz's reagent for the selective removal of acetyl groups from N-acetyl purine and pyrimidine nucleoside analogs
22 was explored by Ferrari et al. in 2015 with 12 examples, resulting in yields of 25-76% for amines
23 (Scheme 8).
11 The compatibility of the Schwartz reagent with different protecting groups was investigated. Amide cleavage was successful for both purine and pyrimidine nucleoside analogs that had various protecting groups, such as OAc (
23a,
23c,
23d, and
23e), OTBDMS, OTHP, OBoc (
23b), OBz, O-trityl and O-isopropylidene groups (
23d). This scope underscores the method's utility in the selective removal of N-acetyl groups in the production of nucleoside-based compounds.
Scheme 8. Deacetylation of protected pyrimidines and purine nucleosides
Nitrone Synthesis from N-Siloxyamides with the Schwartz Reagent
Katahara et al. reported 2017 a reductive methodology for nitrone synthesis, commencing from N-siloxyamides
24 using Schwartz's reagent. Subsequent acid addition yielded functionalized nitrones
25 (Scheme 9).
12 This reaction again exhibited the remarkable chemoselectivity in the presence of a diverse array of sensitive functional groups prone to reduction, such as esters, nitro groups, and olefins. The utility of this methodology was demonstrated in the synthesis and application of functionalized cyclic and macrocyclic nitrones, which were employed for the synthesis of bicyclic isoxazolidines
27,
29, and
31 (Scheme 10) through [3+2] cycloaddition reactions.
Scheme 9. Reductive formation of nitrones from N-siloxyamides
Reduction of Lactams with the Schwartz Reagent
In 2011, Piperno et al. demonstrated the efficacy of the Schwartz reagent for reducing N-alkoxy carbonyl lactams
32, ranging from four to seven-membered rings (Scheme 11).
13 The selective reduction of γ-lactam
32 (n = 1) to lactamol
33 marked a significant advancement in synthetic methodologies. δ-Lactam
32 (n = 2) provided a 4:1 mixture of
33 and enamine
35. β-Lactam
32 (n = 0) and ε-lactam
32 (n = 3) yielded 1:1 mixture of lactamols
33 and aldehydes
34.
Scheme 10. Synthesis of bicyclic isoxazolidines from cyclic nitrones
Scheme 11. Reduction of β, γ, δ, and ε-lactams with the Schwartz reagent
Reduction of Lactams with Cp2Zr(H)Cl Followed by the Addition of Amines and Reductive Amination
In 2019, Prince et al. reported a novel and operationally simple protocol for coupling primary or secondary amines with N-aryl-substituted lactams to produce differentiated diamines with moderate to high yields (Schemes 12 and 13).
14 The process initially involves the reduction of lactams
36 using Schwartz's reagent followed by reductive amination of the aldehyde intermediate with the amine nucleophiles to generate diamines
37. These steps can be combined into a one-pot reaction to streamline the procedure. The methodology's scope was demonstrated with different substituted lactams of various ring sizes to form the desired diamine products, yielding
37a-37j and various primary and secondary amines
37k-
37n. The utility of the reaction was validated by performing gram-scale syntheses. The methodology was extended to include N-aryl pyrrolidinones
38 with enantiopure ester groups, resulting in the formation of α-amino piperidinones
39a-
39j with complete retention of stereochemistry (Scheme 13). The study highlights the utility of lactams as synthons for the synthesis of complex molecules and offers a practical approach to accessing diverse diamine structures. The proposed mechanistic pathway involves a zirconium complex as a masked aldehyde intermediate that, upon reductive amination, is followed by cyclization while retaining stereochemistry. This work opens new avenues for using lactams in organic synthesis and demonstrates the value of innovative reaction strategies for creating complex molecules.
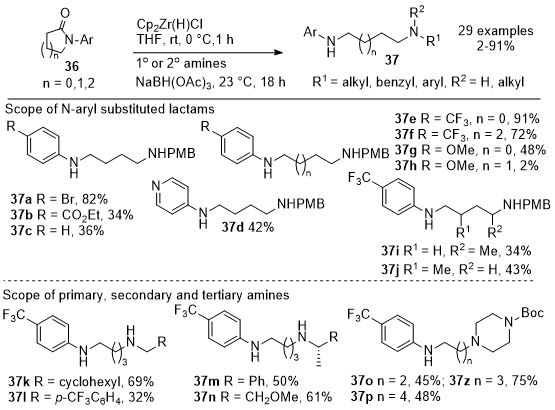
Scheme 12. Aryl-substituted lactams and amines in the one-pot reductive coupling reaction
Scheme 13. Reductive coupling/cyclization sequence of enantiopure N-arylated pyrrolidinones
Chemoselective Reductive Alkylation of Amides and N-Methoxy Amides to Form α-Substituted Amines
Oda et al. reported in 2012 the direct allylation of amides
40 and
42 with allyltributylstannane, resulting in the formation of either substituted tertiary amines
41 or secondary amines
43 using Schwartz's reagent (Scheme 14).
15 Notably, the need for a pre-activation step, typically required to enhance the electrophilicity of amides, was avoided. This method facilitates the direct functionalization of amide groups without additional functional group support. This reaction displayed significant tolerance towards various functional groups, proceeding smoothly even in the presence of electrophilic and other sensitive groups. Tertiary amides formed allylated tertiary amines
41a-41h, and secondary amides formed allylated secondary amines
43a-43f. In 2014, Nakajima et al. extended the direct allylation chemistry to tertiary amides
44 to form tertiary α-allyl amines
45 (Scheme 15).
16 Having established chemoselective reductive nucleophilic addition to tertiary amides, their focus shifted to secondary amides
46, which after reaction with allylzinc bromide yielded secondary amines
47a-47g. In this report,
16 they further extended their work to N-methoxy amides
48, significantly broadening the scope of their work, which yielded significantly improved outcomes compared to tertiary and secondary amides in both yield and chemoselectivity when utilizing a catalytic amount of Sc(OTf)
3 for the synthesis of
49a-49h (Scheme 16). The reaction allowed them to use different nucleophiles, such as indole, enol ethers, TMSCN, and tributyl(propa-1,2-dien-1-yl)stannane, while maintaining the high chemoselectivity for both tertiary amides
50 to generate amines
51a-51g and N-methoxy amides
52 to generate N-methoxy amines
53a-53g (Scheme 17).
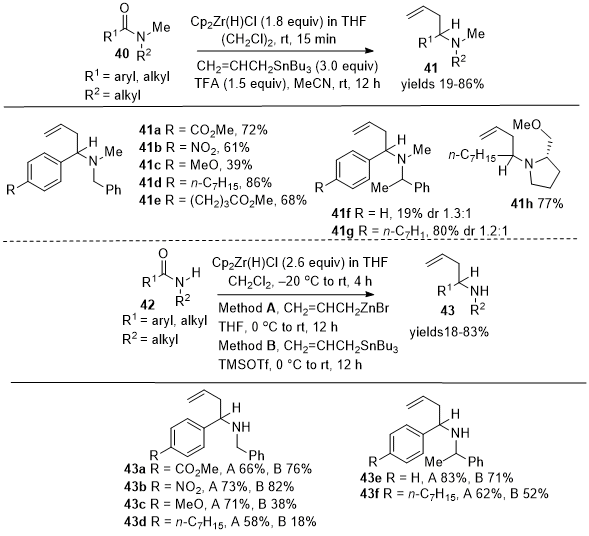
Scheme 14. Reductive allylation of tertiary and secondary amides with allyltributylstannane
Scheme 15. Reductive allylation of tertiary and secondary amides with allyltributylstannane and allylzinc bromide
Scheme 16. Reductive allylation of N-methoxy amides with allyltributylstannane
Scheme 17. Reductive addition of carbon nucleophiles to tert-amides and tert-N-methoxamides
In 2014, Szcześniak et al. introduced a procedure involving using Schwartz's reagent to treat sugar-derived lactams
54, forming the corresponding imines in excellent yields.
17 However, the isolation and purification of these cyclic imines were found to be challenging due to their inherent instability. To address the instability issue, a one-pot protocol was developed, by treating the crude imine solution with Yb(OTf)
3 followed by the addition of allyl tributylstannane (Scheme 18). This approach yielded a mixture of diastereomeric homoallylic amines
55a, and
55e-
55g in good yields (55-91%) and moderate to good stereoselectivity. The allyl tributyltin addition occurred syn to the BnO substituent at the C3 position for six-membered imines. In contrast, steric effects controlled the nucleophile addition for five-membered imines, leading to an anti-arrangement of the BnO at C3 and the allyl group at C2. Further exploration included testing other nucleophiles, TMSCN (
55b and
55h), PhMgBr (
55c and
55i), and the TMS-enol ether of acetophenone (
55d and
55j) yielding cyclic amines in moderate to good yields and selectivities.
17 This versatile protocol was applied to γ-lactam
56 to synthesize two pyrrolidine derivatives, 6-deoxy-DMDP (
57) and radicamine B (
58).
17 Overall, this method enables direct nucleophile addition to
in situ generated cyclic imines, offering opportunities for synthesizing various polyhydroxylated pyrrolidines and piperidines, valuable in natural product synthesis and biosynthetic pathways.
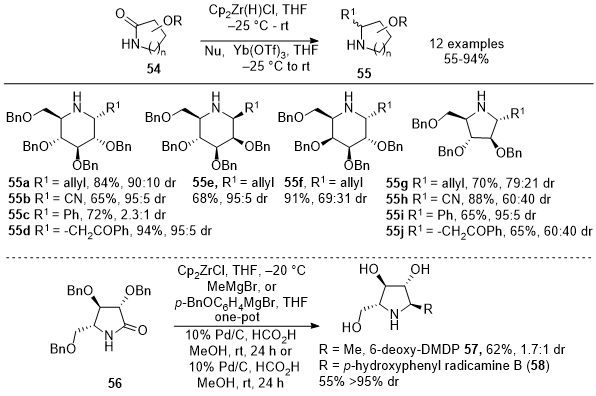
Scheme 18. One-pot reduction of sugar-derived lactams with Schwartz's reagent followed by nucleophilic addition of carbon nucleophiles
In 2017, Zheng et al. reported an effective chemoselective C-C bond method for the one-pot transformations of amides into different compound classes (Scheme 19).
18 They demonstrated that the reductive addition of isocyanoacetates
59 to amides and lactams yields 5-methoxyoxazoles
58 and bicyclic imidazolines
60. This procedure involves partial reduction of amides with Schwartz's reagent, followed by selective addition of the carbon from isocyanide
59. The method was efficient for synthesizing 5-methoxyoxazoles such as
58a-58b from tertiary amides with various alkyl groups. The reaction was also extended to heteroaromatic amides, yielding the corresponding oxazoles such as
58d. The authors also investigated secondary lactams, discovering that 2.2 equiv of Schwartz's reagent were needed in the reaction with isocyanoacetates to produce oxazoles such as
58i. The reaction of isocyanoacetates with 2-pyrrolidines gave separable diastereomeric mixtures of bicyclic imidazolines
60a-60e in excellent yields. However, six-membered lactams provided low yields of compounds such as
60f.
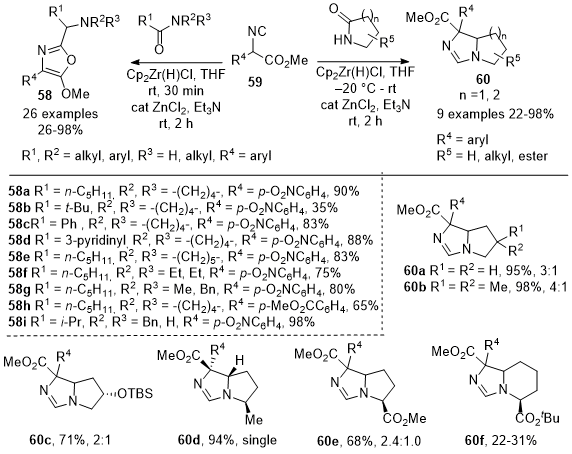
Scheme 19. One-pot reductive nucleophilic addition of methyl-2-isocyanoacetates 59
Ulikowski and Furman reported in 2016 the synthesis of 2,3-disubstituted indoles
62 starting from 3-substituted oxindoles
61 (Scheme 20).
19 The approach leveraged the unique reactivity of Schwartz's reagent, specifically its ability to selectively activate amide carbonyls. The reactive iminium intermediate enabled the addition of diverse nucleophiles, followed by partial reduction, all in a one-pot process, resulting in the formation of 2,3-disubstituted indoles
62. The reaction with nucleophiles such as allyl tributyl stannane (
62a-62b), acetophenone enol TMS ethers (
62c-62d), and indole (
62e-62f) provided the desired products in good yields. One equivalent of the thiophenol is sufficient to form
62g-62h and to avoid over-reduction. An activating group such as TMSOTf is required for dimethyl malonates to provide the desired indoles
62i-62j in good to excellent yields. This method holds promise for synthesizing indole derivatives with pharmacological and synthetic relevance.
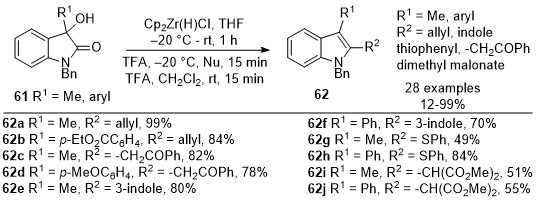
Scheme 20. Synthesis of indoles from oxindoles
Synthesis of 2-Fluoroalkyl Amines from Fluoroacetamides
In 2019, Czerwinski and Furman reported the reductive addition of nucleophiles to fluoroacetamides
63 to synthesize functionalized amines
64 using secondary fluoroamides as a replacement for fluorinated aldehydes (Scheme 21).
20 Model studies involving 2,2,2-trifluoro-N-phenylacetamide and indole as the nucleophile identified the Schwartz reagent as the only reducing agent that would produce the intended functionalized secondary amines
64 in good to moderate yields. The methodology was applied to synthesize the difluoromethyl analog
64c and the heptafluoropropyl analog
64d in satisfactory yields. The functionalization of the intermediate imines by reaction with a diverse range of nucleophiles was performed using the established conditions to synthesize compounds
66a-
66f from
65 as depicted in Scheme 21. The methodology provided access to trifluoromethyl bioisosteres of important drugs, namely the antiarrhythmic procainamide
68 from
67 and prokinetic itopride
70 from
69, as shown in Scheme 22.
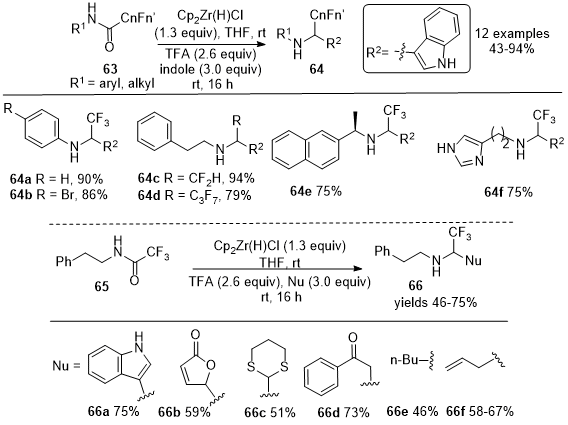
Scheme 21. Directed functionalization of fluoroacetic acid-derived amides
Scheme 22. Synthesis of trifluoromethyl bioisosteres of antiarrhythmic procainamide and prokinetic itopride
In their 2022 study, Tran et al. described an efficient method for synthesizing 1-C-phosphonomethyl and 1-C-phosphonodifluoromethyl iminosugars
72 using sugar-derived lactams
71 (Scheme 23).
21 Using Schwartz's reagent, this process employs a one-pot reaction, forming imines from iminosugars, after which LiCH
2P(O)(OEt)
2 and LiCF
2P(O)(OEt)
2 were added to produce glycosyl phosphonates
72. The yield of this reaction, which was as high a 64%, is influenced by the configuration and the protecting groups present in the sugar lactams, and the reaction proceeds with notable stereoselectivity. The iminosugars synthesized via this method exhibit promising characteristics as transition state inhibitors of glycosyltransferases. Their potential arises from the more stable P-C bond, offering an advantage over the naturally occurring, more hydrolysable P-O bond.
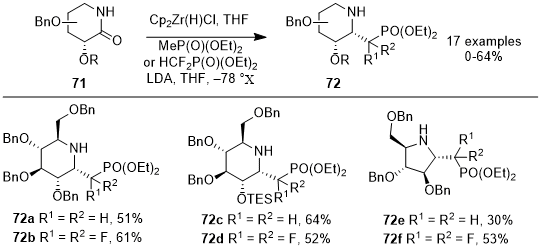
Scheme 23. Reductive alkylative synthesis of 1-C-phosphonomethyl and 1-C-difluoromethyl iminosugars from sugar-derived lactams using the Schwartz reagent
Synthesis of α-Amino Phosphonates from Amides
In 2013, Gao
et al. reported the first method for the reductive phosphination of amides
73 using Schwartz's reagent in a single step (Scheme 24).
22 This reaction method introduces an innovative pathway to obtain α-amino phosphonates
74 after the reaction of amides
73 with the Schwartz reagent and diethylphosphonate, with yields ranging from good to excellent. These reactions operate effectively under mild conditions with a many substrates, including secondary and tertiary amides. Various secondary amides, such as aryl, alkyl, and alkenyl amides were converted into the corresponding α-amino phosphonates
74 in yields ranging from good to excellent. They observed that amides with a hydroxyl group were not converted to product. This could be attributed to the deactivation of Cp
2Zr(H)Cl by the available hydrogen species. The reductive phosphorylation of tertiary amides led to the formation of tertiary α-aminophosphonates such as
74a. H-phosphonates starting materials containing diphenyl, diisopropyl, dimethyl, and acetal groups were used to this reductive phosphination reaction to furnish analogs such as
74e and
74f. The reactivity of the dialkyl phosphonates remained largely unaffected by the specific alkyl moieties.
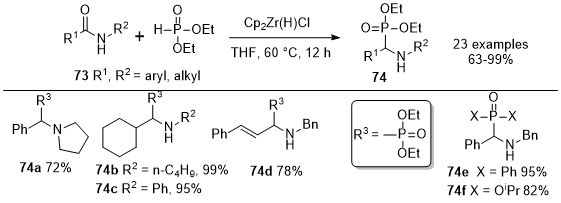
Scheme 24. Transformation of secondary and tertiary amides into α-amino phosphonates
In 2015, Szcześniak et al. reported a direct and efficient approach for synthesizing polyhydroxylated piperidine and pyrrolidine peptidomimetics as outlined in Scheme 25.
23 This strategy involves a one-pot reduction of sugar-derived lactams
75 using Schwartz's reagent, followed by a multicomponent Ugi-Joullié reaction. Chiral lactams
75 were treated with Cp
2Zr(H)Cl, yielding the corresponding imine
76, which was then subjected to a Joullié-Ugi reaction by adding TFA and isocyanide. This process proceeds smoothly with aliphatic (
tBu, Cy) and aromatic (PMP) isocyanides, providing products
78 with up to 95:5 dr. This approach facilitates not only the synthesis of proline amides but also pipecolic acid amides in a one-pot method, enhancing the overall scope of this synthetic method.
Scheme 25. Sequential lactam reduction/Joullié-Ugi three-component reaction
In 2014, Szcześniak et al. introduced a direct and efficient method for synthesizing quinolizidine such as
80a and
80b and indolizidine such as
80c and
80d from iminosugars
79 (Scheme 26).
24 This innovative approach involves a one-pot reduction of sugar-derived lactams
79 using Schwartz's reagent, followed by a diastereoselective Mannich/Michael tandem reaction with Danishefsky's diene. Initially, lactams are treated with Cp
2Zr(H)Cl (1.6 equiv) in THF, forming the corresponding imine. The resulting imine is then subjected to cyclocondensation with the diene and Yb(OTf)
3 within the same reaction vessel. This process provided good yields (51-81%) of bicyclic enaminones
80 and good to high diastereoselectivities of up to 98:2 dr, making this method a valuable tool for efficiently synthesizing these complex natural product scaffolds.
Scheme 26. Synthesis of indolizidines and quinolizidines via one-pot reduction/Mannich/Michael tandem reaction
Applications of the Chemoselective Conversion of Amides to Aldehydes
Summary: In addition to the methodology development described above, chemoselective conversion of amides to aldehydes using Schwartz's reagent was used to generate intermediates within methodology studies
25,26,27 and also employed to generate aldehydes during the total synthesis of natural products.
29,30,31,32,33,34,35,36,37,38,39 The impressive breadth of application demonstrates that the method is an important tool for synthetic organic chemistry.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved