Org. Synth. 2024, 101, 242-257
DOI: 10.15227/orgsyn.101.0242
Preparation of 2-Chloro-1-(1-ethoxyvinyl)pyridinium Triflate
Submitted by John P. Frank, William R. Strutton, Joel K. A. Adade, and Max M. Majireck*
1Checked by Christopher C. Nawrat and Kevin Campos
1. Procedure (Note 1)
A. 2-Chloro-1-(1-ethoxyvinyl)pyridinium Triflate (1). An oven-dried 250 mL three-necked round-bottomed flask (24/40) containing an egg-shaped Teflon-coated magnetic stir bar (32 mm x 15 mm) is fitted with a 60 mL pressure-equalized addition funnel (24/40) and gas-adapter (24/40) connected to a Schlenck line. Rubber septa are added to the top of the addition funnel (24/40) and the remaining side neck (24/40) of the three-necked round-bottomed flask (Figure 1A) (Note 2).
2-Chloropyridine (3.50 mL, 36.6 mmol, 1.1 equiv) (
Note 3), 40 wt. %
ethoxyacetylene in hexanes (
8.77 mL, 36.6 mmol, 1.1 equiv) (
Note 4), and anhydrous
dichloromethane (40 mL) (
Note 5) are injected via syringe into the three-necked flask through one of the side neck septa. Vigorous magnetic stirring is initiated (
Note 6), and the three-necked flask is cooled in an ice/water bath for at least 10 min. The septum on the addition funnel is briefly removed, and under a flow of inert gas and with the stopcock closed, the contents of a glass ampule of
triflic acid (5 g, 33.3 mmol, 1 equiv) are added through the top of the addition funnel with a glass funnel (Notes
7 and
8). A septum is quickly placed onto the top of the addition funnel. A thermocouple is inserted through the septum of the remaining side neck and used to monitor temperature of the subsequent addition (Figure 1B). Using the stopcock to control the rate of addition, the
triflic acid in the addition funnel is dripped into the three-necked flask over 10 min (
Note 9), producing a dark reddish-brown solution (Figure 1C). Additional
dichloromethane (10 mL) is injected into the top of the addition funnel to rinse the remaining
triflic acid residue off of the funnel's sides, and the stopcock is then opened fully to allow the
dichloromethane and
triflic acid residue to flow into the three-necked flask. The reaction mixture is stirred for 18 h, during which time ice/water gradually warms to a room temperature water bath (23 ℃).

Figure 1. A) Equipment setup; B) Solution of ethoxyacetylene and 2-chloropyridine (photos provided by checkers)
Figure 2. Reaction mixture immediately after the addition of triflic acid (photo provided by checkers)
Figure 3. Crude product prior to recrystallization (photos provided by authors)
At this point, the septum, addition funnel, and gas adaptor are removed from the three-neck flask and the reaction mixture is poured into a 100 mL round-bottomed flask (24/40) and concentrated by rotary evaporation (35 ℃, 100mmHg) to give the crude product 1 as a brown solid (Figure 3). 1,2-Dimethoxyethane (DME, 10 mL) (Note 10) and a rod-shaped stir bar (20 mm x 8 mm) are then added to the 100 mL flask containing the crude product and a condenser is fitted (Note 11). The contents of the flask are heated until the solvent begins to boil and gentle reflux is reached (Figure 4) (Note 12). Once complete dissolution of the solids has been achieved (Note 13), the resulting solution is removed from the heating block and stirred for 1 h, cooling to 23 ℃. (Figure 4B). The flask containing a slurry of product is sealed with a rubber stopper and Parafilm M and placed in a -20 ℃ freezer for 16 h.
The crystalline solid is collected via vacuum filtration on a Büchner funnel fitted with filter paper (Note 14) and washed with cold diethyl ether (5 mL) three times (Figure 4C). The crystals are then transferred to a clean, dry 20 mL scintillation vial and dried under vacuum (0.5 mmHg, 23 ℃) on the Schlenk line (Note 3) for 24 h to yield 2-chloro-1-(1-ethoxyvinyl)pyridinium triflate (7.61 g, 68% yield, 96% purity as determined by qNMR) (Notes 15, 16 and 17) as an orange-brown crystalline material (Figure 4D). If increased purity is desired, the product of the first recrystallization is recrystallized again in the same fashion as before (though only 5-10 mL boiling DME is generally sufficient to dissolve the lesser amount of material resulting from the first recrystallization), giving 2-chloro-1-(1-ethoxyvinyl)pyridinium triflate (6.63 g, 59% yield, 98% purity) (Note 16) as light yellow-tan crystals (Figure 4E) (Notes 18 and 19).

Figure 4. A) Recrystallization set up; B) Crude solid dissolved in hot DME; C/D) Product after first recrystallization; E) Product after second recrystallization (photos A and B provided by checkers and photos C, D, and E provided by authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
trifluoromethanesulfonic acid,
2-chloropyridine,
ethoxyacetylene, hexanes,
dichloromethane,
diethyl ether, and
glyme (
1,2-dimethoxyethane). Additional caution should be taken when using the highly toxic solvent
dichloromethane, and the highly corrosive acid
trifluoromethanesulfonic acid.
2. All glassware is dried in an oven at 120 ℃ for 24 h and assembled while hot using heat-resistant gloves. The apparatus is immediately evacuated and backfilled with
nitrogen before being allowed to cool under an inert atmosphere. The authors dried the apparatus by flame drying using a Benzomatic propane hand torch.
3.
2-Chloropyridine was purchased from Sigma-Aldrich (C69802-100G) and used without further purification.
4.
Ethoxyacetylene solution (~40 wt. % in hexanes) was purchased from Sigma-Aldrich (271365-5G) and used without further purification. Given the volatility of this solution, the use of older, previously opened bottles should be avoided to obtain the best results.
5. Anhydrous
dichloromethane with 40-150 ppm amylene as stabilizer was purchased from Sigma-Aldrich (product # 270997-1L) and used without further purification. The authors report that solvents other than
dichloromethane can be used in the procedure, although the use of these alternative solvents has not undergone checking. See the Discussion section of this manuscript for more details.
6. A magnetic stir plate set to 700 rpm was used.
7. Only fresh and dry
triflic acid, preferably from a sealed ampule, should be used in this procedure. Older, previously opened bottles of
triflic acid that have absorbed water have been found to greatly diminish the yield of this reaction.
Triflic acid (Aldrich, ≥99%, sealed ampule) was used in the procedure.
8. To increase measurement accuracy, the authors weigh the full, sealed ampule of
triflic acid prior to addition. Once emptied, the ampule pieces are re-weighed to calculate the exact amount of
triflic acid added to the solution. In four separate experiments, the exact amount of
triflic acid in the ampule was determined to be 5.02 g, 5.10 g, 5.10 g, and 5.17 g. The slight excess of
triflic acid did not have an observable impact on the reaction outcome.
9. The reaction mixture immediately turns dark reddish-brown upon the addition of
triflic acid. The checkers observed an immediate, moderate exotherm; internal reaction temperature increased from 1 ℃ to 8 ℃ over the course of the ten-minute addition.
10. Although the recrystallization is conducted under air, the checkers used 99.5% anhydrous
1,2-dimethoxyethane, purchased from Sigma Aldrich and used without further purification.
11. The condenser is to contain the vapors from the boiling solvent, which may pose a safety risk. The checkers did not use an inert atmosphere for the recrystallization.
12. The boiling point of
1,2-DME is 85 ℃. Internal temperature monitoring was not used; gentle boiling of the solvent indicates that the desired temperature has been reached.
13. The checkers found that 10 mL was sufficient in both checking runs. If additional
DME is needed, this should be added in small portions of 2-3 mL and the mixture should be allowed to return to reflux before it is judged whether any more is required.
14. The authors used Whatman cellulose filter paper (CFP4, 4.25 cm diameter). The checkers used a disposable CHEMRUS 60 mL plastic filter funnel (CR-1018-40) with a medium porosity frit in place of the Büchner funnel.
15.
2-chloro-1-(1-ethoxyvinyl)pyridinium triflate (
1): mp 103-105 ℃;
1H NMR
pdf (500 MHz, CD
3CN) δ: 8.86 (dd,
J = 6.2, 1.8 Hz, 1H), 8.64 (ddd,
J = 8.4, 7.8, 1.8 Hz, 1H), 8.24 (dt,
J = 8.4, 1.7 Hz, 1H), 8.10 (ddd,
J = 7.8, 6.1, 1.5 Hz, 1H), 4.88 (dd,
J = 5.2, 2.0 Hz, 1H), 4.81 (dd,
J = 5.2, 2.1 Hz, 1H), 4.22 (qd,
J = 7.0, 2.2 Hz, 2H), 1.41 (td,
J = 7.0, 2.1 Hz, 3H);
13C NMR
pdf (126 MHz, CD
3CN) δ: 154.4, 151.1, 148.5, 147.8, 131.5, 127.8, 122.2 (q,
1JCF = 320 Hz,
CF
3), 88.1, 68.7, 14.1;
19F NMR
pdf (470 MHz, CD
3CN) δ: 79.28; HRMS-ES+ (C
9H
11NOCl) calcd. 184.0529 (M+), found 184.0532.
16. The purity of
1 after the first recrystallization was determined to be 96 wt% by qNMR
pdf using using
1,3,5-trimethoxybenzene (Sigma-Aldrich Reagent Plus ≥99%) as the internal standard. The purity of the product obtained after the second recrystallization was determined to be 98% by qNMR
pdf using
1,3,5-trimethoxybenzene as the internal standard.
17. A duplicate reaction on identical scale provided 7.38 g (66%) of the product with 96% purity. A second recrystallization to improve purity was not performed.
18. The melting point (mp) range for twice-recrystallized compound
1 increased to 105-107 ℃.
19. The checkers observed smaller crystals and slightly different colors for the product to those shown in the photos (Figure4 C-E). After one recrystallization, the material was light brown, and after a second recrystallization, the crystals were off-white. Despite these visual differences, the material was analytically identical to that produced by the authors.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Until recently, ketene hemiaminals in which the
N-atom is positively charged have been largely inaccessible to synthetic chemists despite the potential breadth of valuable transformations in which that they can participate.
2,3 In 2018, our laboratory reported the first example of a bench-stable pyridinium ketene hemiaminal,
4 and subsequently synthesized >40 additional analogues in order to establish the accessibility and synthetic versatility of this newer compound class.
5 Toward this end, our most recent goals have been to establish a large-scale synthesis of our most versatile reagent 2-chloro-1-(1-ethoxyvinyl)pyridinium triflate (
1), and extensively evaluate its capacity to engage in a broad range of useful transformations.
6,7 We imagined that the unusual structural features of this compound (an electrophilic pyridinium ring bound to a putatively nucleophilic ketene hemiaminal moiety) could engage in two broad reaction types: A) as a "building block" electrophile for nucleophilic additions or substitutions to the pyridinium ring, and B) as a "trapped-ketenium ion" reagent that can thermally release an
O-alkyl ketenium ion en route to myriad other transformations (Scheme 1).
To date, we have identified multiple directions for the use of
1, most of which are at the proof-of-concept stage but will be the focus of future methodological development. For example, we have demonstrated their use as a "trapped ketenium ion" reagent for acid catalyst-free Friedel-Crafts reactions (e.g.,
1 -->
2, Scheme 2), a modified Mukaiyama salt in peptide couplings (e.g., synthesis of
3), and an electrophilic substrate in exceptionally mild S
NAr reactions with amine nucleophiles (e.g.
1 -->
4).
5,6,7 This latter process has been recently optimized to include a broad range of primary and secondary alkyl and aryl amine nucleophiles, leading to >25 2-aminopyridinium salt products.
7 Removal of the
N-(1-ethoxyvinyl) moiety can be accomplished via, thermal, acidic, or oxidative cleavage yielding 2-aminopyridines in the freebase form
10 (following a basic workup), pyridine HCl-salt
11, or pyridine
N-oxide
12. Notably, 2-aminopyridines are frequent targets in drug discovery,
8,9,10,11 and this method circumvents many issues present in other approaches such as conventional S
NAr that typically employs high temperatures and/or high-boiling solvents that complicate purification
12,13,14,15 or transition-metal catalyzed C-N couplings
16,17,18,19,20,21 that require precious metals, additives, and frequently introduce assay-interfering metal contaminants.
22,23
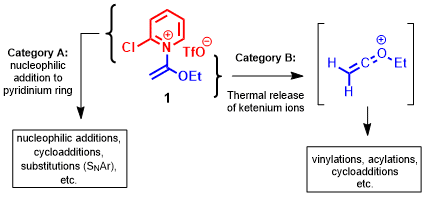
Scheme 1. Potential reaction pathways of pyridinium ketene hemiaminal reagent 1
More recently, we also disclosed preliminary examples of direct, transition metal catalyst-free C3-pyridinylation of indoles (
1-->
7, Scheme 2) and anilines (
1 -->
5).
7 We also extended participating nucleophiles to alcohols and thiols (
1 -->
8 or
9), which undergo S
NAr, and silyl-enol ethers (
1-->
6), which proceed through a C4-addition followed by re-aromatization of the dihydropyridine intermediate. Each of these newer transformations require further optimization, but offer an intriguing glimpse into the versatile range of transformations that can be accomplished with the relatively new bench-stable pyridinium ketene hemiaminal reagent
1. Going forward, we anticipate that provision of gram-scale access to compound
1 will enable our lab and others to significantly expand the repertoire of reaction transformations that this versatile reagent can undergo.
Scheme 2. Preliminary applications of reagent 1 in organic synthesis
As this procedure was being checked, the U.S. Environmental Protection Agency established new prohibitions and workplace restrictions on the use of dichloromethane, making it likely that some research laboratories will be unable use this solvent as described in the above procedure. In light of this, the authors were prompted to examine preliminary dichloromethane-free modifications to the above procedure. In single, unchecked trials performed at the same scale as the checked procedure, the authors observed that using either anhydrous hexanes or anhydrous toluene in place of dichloromethane enabled the formation of compound 1 with moderate to good yield and somewhat lower purity after one recrystallization (Scheme 3). However, it must be strongly emphasized that these additional experiments have not undergone rigorous checking. They merely serve to demonstrate the potential of replacing dichloromethane as the primary solvent, and therefore caution should be taken when attempting any synthetic protocols that deviate significantly from the main procedure detailed above.

Scheme 3. Preliminary results of dichloromethane-free reactions
Appendix
Chemical Abstracts Nomenclature (Registry Number)
2-Chloropyridine; (109-09-1)
40 wt. % Ethoxyacetylene in hexanes; (927-80-0)
Triflic acid: Trifluoromethanesulfonic acid; (1493-13-6)
Glyme: 1,2-Dimethoxyethane; (110-71-4)

|
John Frank graduated from Hamilton College in 2024 with an ACS-certified B.A. in Chemistry. He joined the laboratory of Max Majireck in 2022 to research new synthetic methods enabled by bench-stable pyridinium ketene hemiaminals. This year, he will matriculate to the University of Texas Southwestern Medical Center to pursue a Ph.D. in organic chemistry. |

|
Will Strutton matriculated to Hamilton College in 2022 to pursue a B.A. in Biochemistry/Molecular Biology and Mathematics. He joined the laboratory of Max Majireck in 2022 to research new synthetic methods enabled by bench-stable pyridinium ketene hemiaminals. After graduating from Hamilton College, he intends to pursue an M.D. or M.D./Ph.D. that combines his interests in biochemistry and mathematics. |

|
Joel Adade obtained his B.A. in Biochemistry and Molecular Biology from Hamilton College in 2022 and conducted research in the laboratories of Prof. Wesley Kramer and Prof. Max Majireck. There, he was the recipient of multiple honors and awards including a Posse Foundation full scholarship and the College's highest student honor, the James Soper Merrill Award. Currently, Joel is a research scientist at The Broad Institute's Klarman Cell Observatory and plans to pursue an M.D. or M.D./Ph.D. that combines his interests in research and medicine. |

|
Max Majireck received his B.S. in biochemistry from Grove City College in 2005 while conducting inorganic chemistry research with Prof. Charles Kriley. He received his Ph.D. in 2011 from The Pennsylvania State University while researching natural product total synthesis and organic synthesis methodology in the laboratory of Prof. Steven Weinreb. From 2011-13 he conducted postdoctoral research at Harvard University and The Broad Institute in chemical biology in the laboratory of Prof. Stuart Schreiber. In 2013 he began his independent career at Hamilton College, leading an exclusively undergraduate research program in organic synthesis and chemical biology. |

|
Chris Nawrat was born in Carshalton, England (near London) in 1986. He completed his Ph.D. studies at the University of Nottingham in 2012 working with Professor Chris Moody on several total syntheses. In 2013 he moved to the US to undertake postdoctoral studies in the group of Dave MacMillan at Princeton where he gained expertise in photo- and organocatalysis. Two years later, he joined Merck and Co., Inc. where he enjoys tackling challenging problems in drug development and is currently situated in the Discovery Process Chemistry group based in West Point, Pennsylvania. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved