Org. Synth. 2024, 101, 327-341
DOI: 10.15227/orgsyn.101.0327
Hydration of Nitriles to Primary Amides Enabled by the Ghaffar-Parkins catalyst
Submitted by Daniel W. Turner, Allison T. Hands, and Neil K. Garg*
1Checked by Kohta Ide, Juri Sakata, and Hidetoshi Tokuyama
1. Procedure (Note 1)
A. Benzamide (3). A single-necked (29/42 joint) 250 mL round-bottomed flask is charged with a Teflon-coated magnetic stir bar (2 x 1 cm, football shaped) and the Ghaffar-Parkins catalyst (2) (25.8 mg, 60.1 μmol, 0.002 equiv) (Notes 2 and 3). The flask is then sealed with a rubber septum and pierced with an 18G vent needle and left open to the air.
A separate single-necked (15/25 joint) 100 mL heart-shaped flask is charged with benzonitrile (1) (3.09 mL, 3.12 g, 30.3 mmol, 1.0 equiv) (Note 4). Ethanol (18 mL) (Note 5) and deionized water (12 mL) are then added to the heart-shaped flask using a syringe to give a colorless, clear solution. The benzonitrile solution is then taken up into a 100 mL syringe using a 18G x 20 cm needle and transferred to the 250 mL flask containing the Ghaffar-Parkins catalyst by dispensing the syringe over 1 min. Then the heart-shaped flask is rinsed with ethanol (3 mL) and the solution is transferred to the 250 mL flask with the same syringe (Figure 1A). After repeating the same operation, the rubber septum on the reaction flask is then removed and replaced with a Vigreux distilling column (2.5 cm OD x 35 cm tall, 15/25 upper joint, 29/42 lower joint). The top of the column is fitted with a rubber septum, pierced with an 18G vent needle, and left open to air. The joint between the reaction flask and the column is wrapped tightly with Teflon tape and clamped with a red 29/42 Keck clamp (Figure 1B). The apparatus is then placed in an oil-bath preheated to 80 ℃ and stirred at 300 rpm for 5 h (Figure 1C) (Note 6).

Figure 1. Reaction setup A) after the addition of the benzonitrile solution to the flask containing the catalyst, B) after the assembly of the air condenser, C) 5 h after the addition of benzonitrile (photos provided by checkers)
The reaction flask is removed from the oil bath, and allowed to cool to 23 ℃. The reflux condenser is then detached from the round-bottomed flask, and the stir bar is removed with a magnetic stir bar retriever while rinsing with ethanol (5 mL). The reaction mixture is then concentrated on a rotary evaporator under reduced pressure (40 ℃ water bath, 30 mmHg), yielding a white, crystalline slurry. The crude mixture is then redissolved in methanol (75 mL) (Note 7) and dried over anhydrous Na2SO4 (30 g) (Note 8). The crude reaction mixture is then filtered through a sintered glass funnel into a 500 mL, single-necked recovery flask (Note 9 and 10). The flask containing the filtrate is then charged with silica gel (11 g) (Note 11) and the suspension is concentrated on a rotary evaporator (30 ℃ water bath, 60 mmHg). The flask is then placed under high vacuum (<1.0 mmHg) for 30 min to yield the crude product absorbed onto silica gel.
The product-absorbed silica is then added to a column (4.5 cm OD x 23 cm tall) which is prepared using silica gel (170 g) wetted with 29:1 CH2Cl2:MeOH (450 mL). (Note 12) The flask that contained the product-absorbed silica gel is then rinsed with the eluent (3 x 2 mL), which is subsequently added to the top of the column. Fraction collection (18 x 150 mm culture tubes, 25 mL fractions) begins immediately. The column is eluted with 29:1 CH2Cl2:MeOH (3 L) (Figure 2).
Figure 2. Product absorbed silica dry-loaded onto the silica gel column (photo provided by checkers)
The eluate containing the product (Note 13) is concentrated down on a rotary evaporator under reduced pressure (30 ℃ water bath, 325 mmHg to 150 mmHg). The resulting white solid is dried under high vacuum at 23 ℃ for 30 min to yield benzamide (3) (Figure 3) as a white powder. (3.48-3.53 g, 95-96% yield) (Notes 14 and 15).
Figure 3. Isolated benzamide (3) (photo provided by checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with the
Ghaffar Parkins catalyst (
2),
benzonitrile (
1),
ethanol,
sodium sulfate, silica gel,
methanol, and
dichloromethane.
2.
Ghaffar-Parkins catalyst (
2) was purchased from Strem Chemicals Co. and stored in an argon-filled glovebox to ensure catalyst longevity as suggested by the supplier.
3. Individual reactions can be run open to air. Running the procedure under inert conditions resulted in no impact on yield and purity.
4.
Benzonitrile (99%) was purchased from Tokyo Chemical Industry Co., Ltd. and used as received.
5.
Ethanol (99.5%) was purchased from FUJIFILM Wako Pure Chemical Corporation and used as received.
6. The progress of the reaction is monitored by TLC analysis on silica gel with 29:1
CH2Cl2:
MeOH used as an eluent. The plate is visualized using a UV lamp (254 nm). The
benzonitrile starting material has R
f 0.93 and the
benzamide (
1) product has R
f 0.22 (Figure 4).
Figure 4. TLC of crude reaction mixture (SM = benzonitrile starting material, Co = co-spot of SM and Rx, Rx = reaction mixture with benzamide) (photo provided by checkers)
7.
Methanol (99%, Extra Pure Reagent) was purchased from Nacalai Tesque, Inc. and used as received.
8.
Sodium sulfate (>98.5%) was purchased from Nacalai Tesque, Inc. and used as received.
9.
Methanol (99%, Extra Pure Reagent, 50 mL) was used to wash the reaction flask and the
sodium sulfate to ensure quantitative transfer.
10. Filtration was done using a medium porosity Quark Glass fritted funnel (40-100 μm, 140 mL capacity,) connected to a vacuum pump (Figure 5).
Figure 5. Filtration apparatus (photo provided by checkers)
11. Silica gel (Silica gel 60 N, 0.040-0.050 mm, spherical and neutral) was purchased from Kanto Chemical Co., Inc. and used as received.
12.
Dichloromethane (99.0%, Extra Pure) was purchased from Junsei Chemical Co., Ltd. and used as received.
13. Fractions containing
benzamide 3 were identified using TLC analysis (Figure 6). Using a solvent system of 10:1
CH2Cl2:
MeOH,
benzamide 3 has an R
f = 0.32 and can be visualized under UV light (254 nm). Fractions 27-77 contained the desired product and were collected in a 2 L, single-necked recovery flask. Each fraction was rinsed with
methanol (2 x 3 mL) to ensure quantitative transfer.
Figure 6. TLC analysis showing column fractions (photo provided by checkers)
14. Yields in 3 runs were obtained in 96-97% by checker. The purity of product
3 obtained from chromatography was determined to be 99.2-99.7% by qNMR
pdf using
1,3,5-trimethoxybenzene(Aldrich Chemical Co., Inc., >99.9%) as the internal standard.
15. The product was characterized as follows:
1H NMR
pdf (600 MHz, DMSO-
d6) δ: 8.08 (br s, 1H), 7.92 (d,
J = 7.2 Hz, 2 H), 7.55 (t,
J = 7.8 Hz, 1 H), 7.48 (t,
J = 7.2 Hz, 2 H), 7.40 (br s, 1 H);
13C NMR
pdf (150 MHz, DMSO-
d6) δ: 168.2, 134.5, 131.4, 128.4, 127.7; IR (ATR): 3365, 3166, 1617, 1577, 1448, 1391, 1297, 1122, 769 cm
-1. HRMS (ESI)
m/z [M + H]
+ calcd. for C
7H
8N
1O
1+: 122.0600; found, 122.0602; mp 127.5 - 128.0 ℃.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The accompanying procedure describes a simple and mild means to access primary amides from nitriles. Amides are ubiquitous functional groups utilized across many fields of chemistry and are a prevalent functional group in bioactive molecules.
2 Thus, strategies for accessing amides are of great value to the synthetic community. While primary amides are classically accessed via hydrolysis of nitriles under strongly basic or acidic conditions at elevated temperature, there are two notable drawbacks to employing these forcing conditions. First, it is often difficult to stop additional hydrolysis once reaching the amide functionality, as amide hydrolysis typically occurs at a faster rate than nitrile hydrolysis.
3 Second, such forcing conditions are often not suited to compounds with acid- or base-sensitive functional groups.
4One alternate strategy for the hydration of nitriles leverages transition metal complexes under neutral or mild conditions. Several homogenous and heterogeneous transition metal catalysts for the hydration of nitriles have been reported, as well as enzyme-mediated hydration.
5 Of these catalytic systems, the platinum-based Ghaffar-Parkins catalyst (
2) exhibits high activity and chemoselectivity.
Figure 7: Structure of the Ghaffar-Parkins Catalyst
The Ghaffar-Parkins catalyst (
2) (Figure 7), first disclosed by Ghaffar and Parkins in 1995, has become a popular catalyst for the chemoselective hydration of nitriles under mild conditions.
6,7,8 The Ghaffar-Parkins catalyst consists of a hydride-platinum(II) complex bound to three phosphinite ligands. In the seminal report, the homogenous catalyst exhibited high activity, catalyzing the hydration of acrylonitrile to acrylamide with a turnover frequency of 1485 h
-1.
6 Mechanistically, Ghaffar and Parkins proposed the catalytic cycle begins with protonation of the hydride ligand by water (Figure 8). Subsequent extrusion of molecular hydrogen leaves a vacant coordination site where either a molecule of solvent or substrate can bind. With substrate bound, attack of the hydroxyphospine ligand affords a five-membered intermediate, which is then hydrolyzed to form the amide product and regenerate the cationic active species.
9
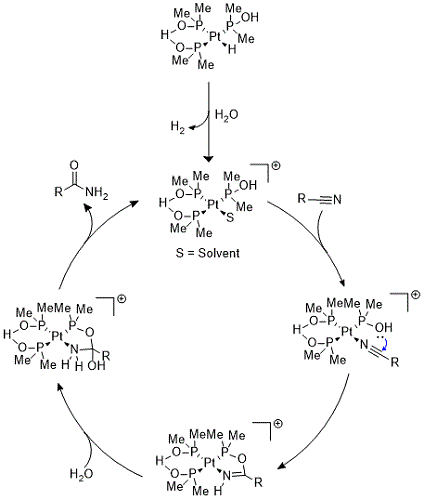
Figure 8: Proposed catalytic cycle for the hydration of nitriles using the Ghaffar-Parkins catalyst
As shown in Figure 9, the platinum-catalyzed reaction conditions are tolerant of nitrile derivatives with diverse steric and electronic character. One particularly difficult transformation for previous methodologies involves the hydration of sterically hindered nitriles.
10 The Ghaffar-Parkins catalyst was able to overcome this limitation and hydrate tertiary nitriles appended to a cyclopropyl ring and a
t-butyl group (
4 &
5), respectively.
4 Additionally, acid- or base- sensitive groups were unaffected by the mild reaction conditions. Specifically, the acid-sensitive 2-furonitrile was hydrated to
6 in 95% yield, and d-amygdalin was converted to amide
7 without epimerization of any of its stereogenic centers.
4 One limitation of the Ghaffar-Parkins catalyst is its inability to efficiently hydrate cyanohydrins. HCN generated from the dissociation of cyanohydrins is proposed to irreversibly bind to the vacant coordination site and shut down catalysis.
11
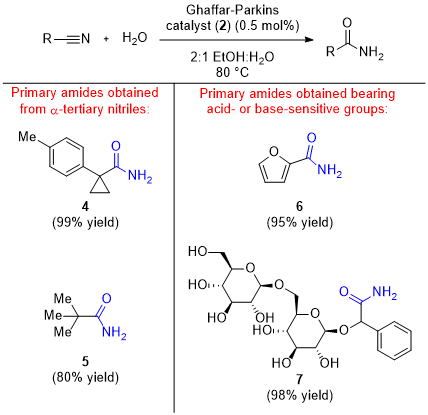
Figure 9: Examples of amide products accessible from nitriles using catalyst (2)
The Ghaffar-Parkins catalyst (
2) has also seen great use in total synthesis studies, including late-stage usage (Figure 10). One of the first synthetic applications of the Ghaffar-Parkins catalyst was in the synthesis of atenolol. Using only 0.0001 mol% catalyst loading, Parkins and colleagues were able to access
8 in the presence of an unprotected secondary alcohol and amine.
12 In route towards the synthesis of an anticancer alkaloid stephacidin B, Myers and coworkers employed 20 mol% Ghaffar-Parkins catalyst to access primary amide
9.
13 This amide was further elaborated to a lactam present in the natural product. Finally, in a formal synthesis of diazonamide A, Sammakia and co-workers reported the formation of
10 in 92% yield from the corresponding nitrile utilizing only 2 mol% Ghaffar-Parkins catalyst. This amide was later cyclized to form an oxazole present in the natural product.
14Figure 10: Applications of Ghaffar-Parkins Catalyst in Total Synthesis
In summary, this procedure provides a facile approach to access primary amides from nitrile precursors utilizing the Ghaffar-Parkins catalyst. The mild reaction conditions tolerate a variety of functional groups that are vulnerable under more classic hydrolysis methods. Applications in total synthesis have already shown this methodology to be a versatile tool to synthesize amides in complex settings.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Benzonitrile (1) (100-47-0)
Ghaffar-Parkins Catalyst (2) (173416-05-2)

|
Daniel Turner was born and raised in San Diego, California. In 2022, he received a B.S. in Biochemistry from the University of California, Los Angeles where he performed research in the lab of Professor Ellen Sletten. In 2022, he continued with graduate studies at the University of California, Los Angeles, where he is currently a second-year graduate student in Professor Neil K. Garg's laboratory. His studies primarily focus on developing synthetic methods involving strained cyclic intermediates. |

|
Allison Hands was born in Los Angeles, CA and raised in Barrington, RI. In 2019, she received her B.A. in Chemistry with a concentration in Biochemistry from Skidmore College where she performed research in the lab of Professor Steven Frey. Between 2019 and 2022, she worked as a medicinal chemistry research associate at the Broad Institute in Cambridge, MA. Then, in 2022, she moved to the University of California, Los Angeles, where she is currently a second-year graduate student in Professor Neil Garg's laboratory. Her studies primarily focus on developing synthetic methods utilizing strained cyclic allenes. |

|
Neil Garg is the Distinguished Kenneth Trueblood Professor of Chemistry at the University of California, Los Angeles. His laboratory develops novel synthetic strategies and methodologies to enable the total synthesis of complex bioactive molecules. |

|
Kohta Ide was born in Sapporo, Japan in 1997, and received his BSc (2020), MSc (2022) from Tohoku University under the supervision of Professor Hidetoshi Tokuyama. He is currently pursuing his Ph.D. at the same graduate school. His research interests are the area of the total synthesis of complex natural products. |

|
Juri Sakata was born in Shizuoka, Japan in 1986, and received his BSc (2009), MSc (2011) from Kogakuin University under the supervision of Professor Shinji Nagumo and Professor Masaaki Miyashita. He then moved to the laboratories of Professor Keisuke Suzuki at the Tokyo Institute of Technology and got his Ph. D. in 2015. In 2015, he joined the group of Professor Hidetoshi Tokuyama and was appointed Assistant Professor. His current research interest is total synthesis of complex natural product. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved