Amides are omnipresent in organic and medicinal chemistries as well as material, polymer and crop sciences.
1,2 Typically, the amide bond is formed through the condensation of an activated carboxylic acid function and a free amine group. Numerous activating reagents for the acid moiety have been designed and developed to avoid racemization problems and tolerate encumbered and less reactive substrates.
3,4 However, to combine all these requirements using cheap, stable, and safe activating reagents in an atom-economy manner, is still a challenge. These points justify ongoing, assiduous research in this field. Our group has been interested on developing alternative ways to synthesize the amide bond, mainly for peptide chemistry purposes.
5 Traditionally, chemical syntheses of peptides, either in solution or solid phase (SPPS), are carried out from the C→N direction by using the classical carboxylic acid activation-amine condensation reaction sequence.
3,4 Nevertheless, the ribosomal synthesis of peptides and proteins takes place from the opposite N→C direction. Contrary to the "natural" N→C direction syntheses, C→N conventional strategies warrants racemization-free processes (or at least very minimized ones).
6 However, C→N direction peptide synthesis involves various group protection/deprotection manipulations and large excess of non-recoverable activating reagents that consequently delivers waste.
In this context, we started a research program seeking to provide a practical way to reach amides, for small-peptide synthesis, through the challenging and underdeveloped N→C direction.
7,8,9,10,11,12 In 2014, we described the use of CDI-protected α-amino esters (CDI,
e.g.,
N,N'-carbonyldiimidazole) in inverse peptide synthesis.
13 The procedure involves the coupling of activated amine partners instead of usual carboxylic activation and has been illustrated in
Organic Syntheses one year later (
e.g., Scheme 1).
14 CDI-activated α-amino esters have unique features that affect the reactivity of the native α-amino acids towards amide bond formation. Indeed, during their coupling reaction with another amino acid partner, bearing a free carboxylic acid function, they can act as masked isocyanate intermediates that are prone to nucleophilic attack by the carboxylate (see below on mechanism discussion).
15 The CDI-mediated synthesis of activated α-amino esters was described, in the general procedure, by reaction of amino ester substrates with equimolar amounts of both CDI and DIPEA in CH
2Cl
2 at room temperature over 12-24 h. The resulting activated amino esters were isolated by filtration over silica gel and stored for months at 4 ℃ without degradation. Subsequently, they were exposed to coupling reactions with N-protected (
e.g., Fmoc, Boc, Cbz) α-amino acids (0.67 equiv) in the presence of 10 mol% of CuBr
2 and HOBt in CH
2Cl
2 (1 M) at room temperature over 20 h. It is important to mention that peptide bond formation using CDI-protected amino esters takes places in the absence of an added base, which is quite unusual in peptide couplings. Flash column chromatography on silica gel furnishes the required dipeptides in high yields without detected racemization even when a sensitive cysteine residue is engaged.
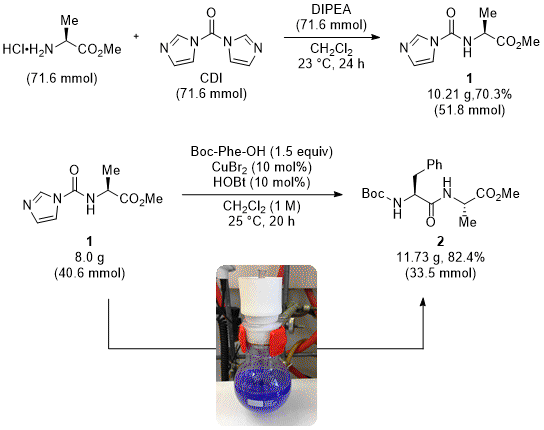
Scheme 1. Dipeptide synthesis via activated α-amino esters
13,14Unlike traditional peptide synthesis, the originality of this work lies in the possibility of iteratively elongating the chain in the inverse N→C direction via the activation of amine functions without the need for a base. Indeed, whereas the use of CDI to activate the carboxylic acid function is well known and reported in the literature, its use to activate the amine function in traditional peptide synthesis was unprecedented.
In this Discussion Addendum we describe experimental improvements since the publication of the first communication on the subject as well as the transposition of the method to the synthesis of general amides.
Experimental Developments
Sequential one-pot dipeptide synthesis
If the activation of the free-amine function is the key feature for the preparation of short-peptide sequences in inverse N→C direction in a base-free procedure, the requirement for the synthesis and purification of the CDI-activated intermediates detracts from its attractiveness. Indeed, in traditional peptide synthesis, carboxylic acids are
in situ activated with coupling reagents and are not isolated. To circumvent the need for isolating the activated amino esters, in 2017 we have developed a sequential one-pot strategy for the preparation of dipeptides (Scheme 2).
16 The more practical procedure combines the
in situ amine activation in the presence of CDI (1.5 equiv) in CH
2Cl
2 at room temperature for 30 min. One side-compound of this step is imidazolium salt (HCl·imidazole,
3) that in some cases precipitated from the reaction medium. This observation was very practical as the
in situ activation could be visually monitored, otherwise followed by TLC. Thus, the coupling reaction step takes place by addition of the second amino acid partner and catalytic amounts of both CuBr
2 and HOBt (10 mol%) into the flask containing the freshly prepared CDI-activated intermediate. After additional 20 h, dipeptides were isolated after acid-base extraction followed by filtration on a short pad of silica gel.
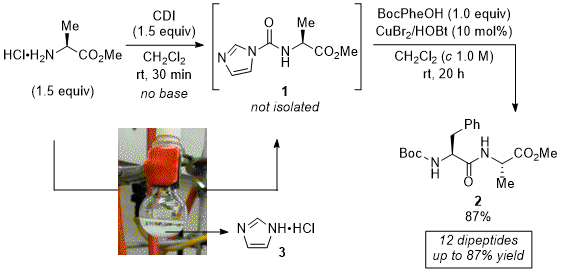
Scheme 2. Sequential one-pot dipeptide synthesis via activated α-amino esters
16Overall, yields are slightly improved or similar to the ones using the former procedure. In addition, dipeptides are isolated without detected epimerization. However, contrary to the stepwise protocol, no base is required for the CDI activation step. Indeed, base addition during the CDI-activation stage almost completely inhibits the sequential synthesis of dipeptide, resulting in barely trace amounts of the expected products (7
vs 87% yield). This observation corroborates past studies, which have shown that the addition of a base adversely affects amide formation.
13 In this amended procedure, the activated amine intermediates are
in situ generated and their isolation can be bypassed providing an easier and less wasteful alternative.
Microwave irradiation or conventional heating for dipeptide synthesis via CDI-activated α-amino esters
With a sequential one-pot strategy developed, the use of CDI-base α-amino ester in peptide synthesis can be compared, in terms of reaction conditions, to the traditional peptide synthesis (where activated carboxylic acid intermediates are not isolated). Nevertheless, the procedure has the advantage of providing a more practical, less cost-effective and challenging non-conventional (
e.g., N→C direction) method. However, to become a common, routine synthetic alternative in peptide synthesis, the reaction kinetics need to be reworked and considerably improved. Indeed, either in the stepwise method or in the sequential one-pot strategy, coupling reactions take place over 20 h. Consequently, next improvements on the method were focused on reducing the time of the coupling reaction step. Several activating agents for the amine function, and numerous solvents and additives have been surveyed. Still, none provided better results than the initial combination (
e.g., CDI, CuBr
2/HOBt, CH
2Cl
2).
17In contrast, when the reactions were run under microwave irradiation or conventional heating (
e.g., 60 ℃), a notable improvement on the overall reaction time for the coupling reactions was observed (Scheme 3). Indeed, the time was reduced from 20 h to only 30 min.
17 Also, no racemization has been detected and dipeptides were isolated in comparable or slightly improved yields either under microwave or conventional heating.
Scheme 3. Microwave or conventional heating for dipeptide synthesis via activated α-amino esters
17Mechanism insights
In parallel to these experimental improvements, mechanistic studies based on experimental observations and NMR monitoring were carried out.
17 It is believed that a carbamic anhydride intermediate
4 is a key intermediate that provides amides after CO
2 extrusion
18 (Scheme 4). This intermediate might be formed either via direct addition of the carboxylate into the CDI-protected α-amino esters or via the formation of an isocyanate
515 through the loss of imidazole. Experimental results, such as the absence of racemization during the coupling reactions, associated with precedents in the literature ruled out the possible formation of both oxazolone
6 and acylimidazole
7 as potential intermediates from
4. Moreover, right after the publication of our communication, the reaction mechanism has been investigated by Bi and Jiang using DFT, highlighting that the coupling reactions are more likely to proceed through a decarboxylative 1,3-acyl transfer.
19
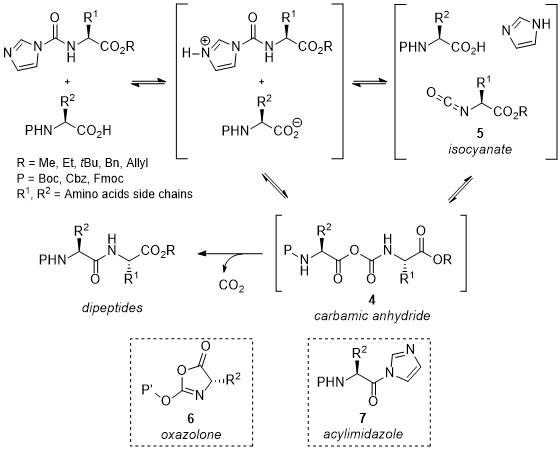
Scheme 4. Mechanism insights for dipeptide synthesis via activated α-amino esters
17In addition, in all developed protocols, scale-up was performed without difficulties and no decrease in yields, compared with the two-step procedure, was observed (Scheme 5). Besides, assessment of residual copper traces through inductively coupled plasma mass spectrometry (ICP-MS) showed an acceptable average of 1.53 ppm of Cu.
20Scheme 5. Large scale syntheses
Applications of the method
Synthesis of a tetrapeptide from N→C direction
As mentioned before, one of the biggest advantages of the method stands on the possibility to achieve peptide synthesis from the inverse N→C direction. However, the major problem associated with this 'natural' peptide elongation concerns epimerization of the C-terminus amino acids stereocenters, which is very hard to control. Within this purpose, this method was highlighted with the racemization-free synthesis of a tetrapeptide model
8 from the challenging inverse N→C direction. Although no specific biological activity is associated to
8, encumbered (Val), unprotected (Trp), functionalized (Met), and prone to racemization (Phe) amino acids were used to validate the inverse method. The coupling reactions take place under the standard conditions.
13 The tetrapeptide was isolated after five linear steps in an iterative racemization-free manner. It is noteworthy to mention that the same inverse strategy using traditional PyBOP as activating agent provided considerable racemization (Scheme 6).
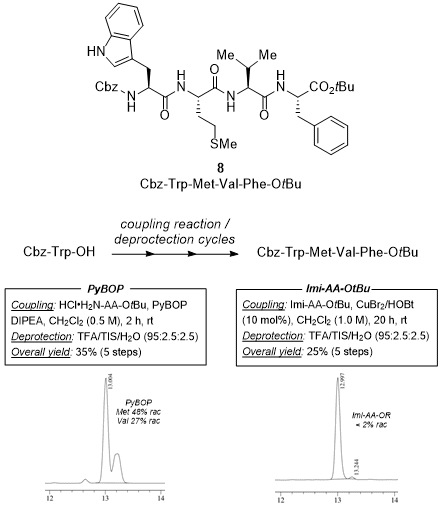
Scheme 6. Iterative synthesis of a tetrapeptide from the inverse N→C direction
13Synthesis of general amides
Since the first publication of our work in 2014 on the use of activated amino esters in small peptide synthesis, improvements in reaction conditions have always been aimed at peptide chemistry. Nevertheless, the ubiquity of the amide function in many other research fields (
e.g., materials, polymers, natural products, hydrogels, medicinal chemistry, …) prompt us to explore the possibility to extend the scope of application of the method to general amides
9 (
e.g., that do not bear amino acid residues). For this purpose, both the stepwise one-pot procedure at room temperature during 20 h and the strategy based on microwave irradiation or conventional heating at 60 ℃ in short reaction periods have proved suitable. Overall, microwave irradiation provided slightly higher yields and better kinetics (60 min
vs 20 h) (Scheme 7). General amides
9 are prepared in good yields. Reluctant, less reactive substrates such as aniline and benzoic acid smoothly react under the reaction conditions to afford amides in moderate yields.
17
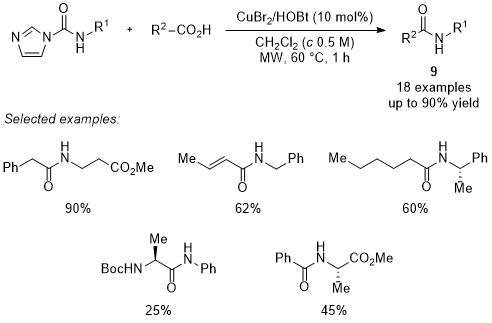
Scheme 7. General amide synthesis via activated α-amino esters
17Synthesis of dipeptides for Self-Induced Diastereomeric Anisochronism (SIDA) studies
Recently, dipeptides synthesized via the sequential one-pot strategy with CDI-activated α-amino esters were used in a study of Self-Induced Diastereomeric Anisochronism (SIDA).
21 This is a quite fascinating phenomenon for chiral recognition process by NMR spectroscopy where enantiomers are able to self-assemble in solution to provide diastereomeric adducts. The objective within this work was to provide a distinctive analytic procedure to accurately determine the enantiomeric composition of scalemic mixtures and survey thermodynamic and stereochemical properties at the basis of SIDA.
In summary, since the publication of our first communication on the use of CDI-activated α-amino esters for inverse peptide synthesis, efforts have been devoted to propose alternative procedures that are still simple, but further practical. CDI is an inexpensive and safer chemical considered as a greener alternative product. Compared to many powerful and widespread activating agents in traditional carboxylic acid activation (
e.g., benzotriazole or azabenzotriazole derivatives), its use allows much safer, atom-saving and cost-effective processes. In addition to these significant advantages, the amine function activation, instead of carboxylic acid, paves the way for an inverse N→C direction peptide synthesis, which is observed in Nature and barely tried in laboratories due to potential racemization issues. A few remarkable alternative methods to the conventional carboxylate activation have been disclosed since 2013. Usually, activated intermediates are not stable and requires
in situ multi-step synthesis and/or the use of toxic reagents. Also, some of these alternative methods are not always compatible with sensitive amino acids. Despite the possibilities opened by the use of CDI-activated α-amino esters in peptide and general amide syntheses,
22 and the advances made to perform coupling reactions in milder and practical operational conditions, further application on peptide chemistry (
e.g., transposition into SSPS) and medicinal chemistry is still required.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved