Org. Synth. 2024, 101, 542-563
DOI: 10.15227/orgsyn.101.0542
Synthesis of 4-(2,2-Difluorovinyl)benzonitrile through a Wittig-type Olefination of 4-Formylbenzonitrile
Submitted by Andrew J. Intelli, Jacob P. Sorrentino, and Ryan A. Altman*
1Checked by Niccolò Carlo Azzini, Christoph Etling, and Dirk Trauner
1. Procedure (Note 1)
A. 4-(2,2-Difluorovinyl)benzonitrile (2). An oven dried 3-neck (24/40 joint) 500-mL round bottom flask is equipped with a 50-mL addition funnel sealed with a rubber septum (24/40), a thermometer adapter (24/40 joint) and thermometer (range -20 - 150 ℃) (Note 2), a 1.5 x 3.2 cm Teflon-coated football-shaped stir bar, triphenylphosphine (19.7 g, 75.0 mmol, 1.50 equiv.) (Note 3) and 4-formylbenzonitrile (6.56 g, 50.0 mmol, 1.00 equiv.) (Note 4) and sealed with a rubber septum (24/40). The flask is then connected to a Schlenk line via a 16-gauge x 1.57 in needle and the system is placed under high vacuum for 15 min (Note 5) before refilling with dry nitrogen, followed by evacuation (1 min) and backfilling the system with dry nitrogen two more times (Figure 1).
Figure 1. Reaction setup under vacuum
Then, dry
DMF (80 mL) (
Note 6) is added to the flask via a 16-gauge x 12 in needle, and the flask is then placed in a heating mantle equipped with sand. The reaction mixture is heated to 90 ℃ (internal temperature) with 800 rpm stirring. Then,
potassium 2-bromo-2,2-difluoroacetate2 (19.2 g, 90.0 mmol, 1.80 equiv.) is measured into a 100-mL heart-shaped flask in a
nitrogen-filled glovebox (Notes
7 through
9) sealed with a 14/20 rubber septum and transferred out of the glovebox. The flask is connected to a Schlenk line under a
nitrogen atmosphere via 16-gauge x 1.57 in needle. To the flask containing
potassium 2-bromo-2,2-difluoroacetate, 40 mL of dry
DMF is added via 16-gauge x 12 in needle. The mixture is swirled by hand under
nitrogen until no visible solids remained (approximately 5 minutes; Figure 2), and then the resulting solution (
Note 10) is transferred via a 60-mL syringe equipped with a 16-gauge x 12 in stainless-steel needle into the 50-mL addition funnel (
Note 11).

Figure 2. Dissolving the bromodifluoroacetate salt in dry DMF
Then, a bubbler is attached via a 16-gauge x 1.57 in stainless-steel needle and plastic tubing to the top of the addition funnel (Note 12), and the solution of DMF/potassium 2-bromo-2,2-difluoroacetate is added dropwise over 10 min to the solution of DMF/triphenylphoshine/4-formylbenzonitrile (Figure 3) (Note 2).
Figure 3. Reaction mixture at 90 ℃ before (left) and after (right) adding the BrCF2CO2K/DMF solution
After the addition is complete, the reaction is allowed to stir for an additional 45 min at 90 ℃ before removing the heating mantle. Then, the crude reaction mixture is allowed to cool to 23 ℃ (Notes 13 and 14). Then, water (120 mL) is added to the crude reaction mixture, followed by Et2O (120 mL) (Note 15). The crude reaction mixture is then transferred to a 500 mL separatory funnel, where the Et2O layer is removed, and the aqueous layer is extracted with Et2O (2 x 60 mL) (Figure 4).
Figure 4. Separatory funnel after transferring the organic and aqueous layers from the 500-mL reaction flask
The organic layers are combined and washed with 10% LiCl in H2O (3 x 40 mL) (Note 16), brine (1 x 40mL) and transferred to a 500 mL Erlenmeyer flask equipped with a 1.5 x 3.2 cm Teflon-coated football-shaped stir bar and dried over 10 g of sodium sulfate (Notes 17 and 18). Methyl iodide (0.81 mL, 13.0 mmol, 1.2 equiv. relative to the remaining PPh3) (Note 19) is added to the organic layer, the flask is sealed with a glass stopper, and the reaction mixture is stirred at 800 rpm for 16 h at 23 ℃. Next, a 150 mL 6.5 in ID x 2.5 in medium-porosity Buchner funnel is placed on top of a 1 L tear-drop shaped flask and is charged with a 6.5 in ID x 0.5 in layer of silica gel (Note 20), followed by a 6.5 in ID x 0.5 in layer of diatomaceous earth (Note 21) (Figure 5).
Figure 5. Silica plug before (left) and after (right) filtration
Hexanes (100 mL) (Note 22) is added to the 500-mL Erlenmeyer flask containing the organic layer, which became turbid from the precipitation of (MePPh3)+I- (Figure 6). The organic layer is vacuum filtered through the pad of silica/diatomaceous earth (Notes 20 and 21) and the pad is washed with Et2O (3 x 100 mL) (Note 23). The filtrate is then purified via column chromatography (dry-loaded using 25 g of diatomaceous earth [Note 21] by mixing with crude reaction mixture and concentrated under reduced pressure) using a 31 cm (length) by 5. 7 (width) column using 208 g silica gel (Note 20) wetted with hexanes (1000 mL; Note 22). The column is then eluted with 300 mL of hexanes (Note 22), followed by 1300 mL of 10% Et2O in hexanes (Note 15, 22 and 24). The fractions are collected in 25 mL test tubes, and the desired product is found in fractions 40-52 (Note 25). These fractions are combined and concentrated under reduced pressure on a rotovap (32 ℃, Note 26).

Figure 6. Before (A), 16 h after methyl iodide addition (B) and after rinsing the flask with diethyl ether (C) isolated product after drying under high vacuum (D)
The resulting colorless crystalline solid is then transferred to a 20 mL scintillation vial and dried under high vacuum for 3 h (Figure 6D) to give a colorless crystalline solid (2.79 g, 16.9 mmol, 34%) (Notes 27, 28, and 29).
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
triphenylphosphine,
benzaldehydes,
potassium 2-bromo-2,2-difluoroacetate,
N,N-dimethylformamide,
diethyl ether, hexanes,
methyl iodide, silica gel, diatomaceous earth,
lithium chloride,
diethyl phosphite and
CDCl3. The use of a bubbler when using this procedure is required to prevent over pressurization of the reaction set-up due to the decarboxylation of
potassium 2-bromo-2,2-difluoroacetate. The exothermic nature of the reaction requires the use of an internal thermometer to monitor the increase in temperature.
2. The addition of the
potassium 2-bromo-2,2-difluoroacetate solution causes an observable increase in temperature measured by the internal thermometer. The rate of addition was carefully controlled to maintain a reaction mixture temperature under 100 ℃.
3. The authors used
triphenylphosphine (99%) purchased from Oakwood Chemicals, which was used as received. The checkers used
triphenylphosphine (99%) purchased from Thermo Scientific Chemicals and used the chemical as received.
4. The authors used
4-formyl benzonitrile (97%) purchased from Combi-Blocks, which was used as received. The checkers used
4-formyl benzonitrile (97%) purchased from Ambeed and used the chemical as received.
5. Placing
PPh3 under high vacuum for 15 min helps to partially dry the reagent.
6. The authors used
NN-dimethylformamide (> 99.8%) purchased from Fisher Scientific (Certified ACS). It was passed through two columns of activated molecular sieves under argon in a JC Meyers Phoenix Solvent Dispensing System before use. The checkers used
N,N-dimethylformamide (99.8%, Extra Dry, under an AcroSeal) from Thermo Scientific Chemicals. The chemical was used as received from the bottle.
7. The checkers used
potassium 2-bromo-2,2-difluoroacetate (95%) from Ambeed. The chemical was used as supplied.
8.
Potassium 2-bromo-2,2-difluoroacetate was stored in a
nitrogen-filled glovebox due to the hygroscopicity of the salt. In place of a
nitrogen-filled glovebox, a dry box desiccator can be used.
9. The checkers stored
potassium 2-bromo-2,2-difluoroacetate in a desiccator over Drierite under argon and handled the compound without a glovebox. During the checkers' runs, the compound was weighed quickly under air.
10. The checkers were not able to completely dissolve the compound in the
DMF during both checking runs. The compound was therefore added as a partially dissolved suspension which did not impact the yield of the reaction. Overall volume of
DMF was not increased.
11. The
potassium 2-bromo-2,2-difluoroacetate in
DMF mixture should be made immediately prior to use as
potassium 2-bromo-2,2-difluoroacetate slowly decomposes in
DMF to produce a yellow color (Figure 7). However, no differences in yield and purity are observable if the stock solution is used within 1 h, even if yellowing has occurred.
Figure 7. Decomposition of 2-bromo-2,2-difluoroacetate in DMF
12. The reaction proceeds with significant evolution of gas and a silicone oil bubbler should be used to ensure proper venting of excess pressure from the system.
13. If desired,
31P NMR can be utilized at this point to determine the amount of unreacted
PPh3 remaining in the crude reaction mixture. This determination allows for the use of a minimal amount of MeI to quench the residual
PPh3. For this preparation,
diethyl phosphite (129 μL, 1.0 mmol, 7.8 ppm) was added to the crude reaction mixture as an internal standard and thoroughly mixed at 800 rpm for 5 min before taking 0.5 mL of crude reaction mixture for
31P NMR analysis.
Diethyl phosphite was integrated as 1.0 (7.8 ppm), and
PPh3 was found to integrate to 11.1 (11.1 mmol; -6.1 ppm). Then 1.2 equiv. of MeI relative to the remaining
PPh3 was used to quench the residual
PPh3.
14. The authors used
diethyl phosphite (98%) purchased from Acros Organics, which was used as received. The checkers purchased
diethyl phosphite (98%) from Sigma-Aldrich and used the chemical as received.
15. The authors used
diethyl ether (anhydrous, ACS grade, BHT stabilized) purchased from Fisher Scientific, which was used as received. The checkers purchased
diethyl ether (anhydrous, ACS grade, BHT stabilized) from Oakwood Chemicals and used the solvent as received.
16. The authors used
lithium chloride (99%, anhydrous) purchased from Oakwood Chemicals, which was used as received. The checkers purchased
lithium chloride (98%+, anhydrous) from Thomas Scientific and used the chemical as received.
17. The authors used
sodium sulfate (99.5%, anhydrous) purchased from Oakwood Chemical, which was used as received. The checkers purchased
sodium sulfate (99%, anhydrous) from Thomas Scientific and used the chemical as received.
18. The checkers dried the solution over 10 g of
sodium sulfate, removed the drying agent by filtration through a cotton plug and washed it with 10 mL of
diethyl ether. A stir bar was then added to the obtained dried solution to perform the subsequent alkylation step.
19. The authors used
methyl iodide (99.5%, stored over copper) purchased from Oakwood Chemicals, which was used as received. The checkers purchased
methyl iodide (99+%, stored over copper) from Thermo Scientific and used the chemical as received.
20. The authors used silica gel (SiliaFlash® P60, 40-63 μm, 230-400 mesh) purchased from SiliCycle, which was used as received.
21. The authors used diatomaceous earth (545) purchased from Oakwood Chemicals, which was used as received. The checkers used Celite 545 purchased from Thermo Scientific.
22. The authors used hexanes (>98.5%, certified ACS) purchased from Fisher Scientific, which was used as received.
23. The silica plug was prepared using a 6.5 in ID x 0.5 in layer of silica gel and a 6.5 in ID x 0.5 in layer of diatomaceous earth (Figure 5).
24. Glass-backed silica gel TLC plates (10-12 μm, 60 Å pore size) were purchased from Supelco. Progress of the reaction is monitored by TLC analysis on silica gel plates (Figure 8) using 9:1 hexanes:
diethyl ether as eluent. The plates are visualized using short wave (254 nm) UV light (Figure 8, left).
4-formyl benzonitrile (
1) has R
f 0.09 (UV active), and
4-(2,2-difluorovinyl)benzonitrile (
2) has R
f 0.29 (UV active).
Figure 8. TLC of the reaction mixture after cooling to 23 C developed using 9:1 hexanes:diethyl ether (A = co-spot of aldehyde 1, gem-difluoroalkene 2, triphenylphosphine and crude reaction mixture, B = benzaldehyde, crude = crude reaction mixture, C = triphenylphosphine.
25. Fractions containing the product were identified by TLC analysis using 9:1 hexanes:
diethyl ether as the eluent. The plates are visualized using UV (254 nm). Fractions 40-52 contained the desired product, and each collected test tube was rinsed with (2 x 1 mL)
diethyl ether, to ensure quantitative transfer (Figure 9).
Figure 9. TLC Plates showing column fractions
26. The checkers removed the solvent under reduced pressure using a rotavap; bath temperature: 10 ℃, maximum vacuum applied: 7.5 torr.
27. The purity of product
2 obtained from chromatography was determined to be 98% by qNMR
pdf using
tert-butyl acetate (Thermo Scientifc, 99%) as the internal standard.
28. Characterization data of purified
2:
1H NMR
pdf (400 MHz,
CDCl3) δ 7.62 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 5.34 (dd, J = 25.5, 3.3 Hz, 1H).
13C{
1H} NMR
pdf (101 MHz,
CDCl3) δ 157.1 (dd, J = 301.3, 292.2 Hz), 135.5 (d, J = 7.0 Hz), 132.5, 128.2 (dd, J = 6.9, 3.6 Hz), 118.8, 110.7 (t, J = 2.4 Hz), 81.9 (dd, J = 30.5, 12.9 Hz).
19F NMR
pdf (376 MHz,
CDCl3) δ -77.80 (d,
J = 20.5 Hz), -79.46 (d,
J = 20.5 Hz). IR (neat): 3043, 2971, 2227, 1730, 1688, 1610, 1514, 1415, 1371, 1325, 1284, 1243, 1172, 946, 855, 832, 820, 601, 548, 512cm
-1. HRMS-APCI (
m/
z) [M + Na]
+ calcd for C
9H
5F
2NNa, 188.0282; found, 188.0280; mp 66-67 C; R
f (9:1 hexanes:
Et2O) 0.29.
29. A second run on half-scale carried out by the Checkers provided
2 in 38% yield with 97% purity by qNMR.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Fluorinated functional groups perturb physiochemical and pharmacological characteristics of compounds, and have garnered the attention of synthetic, pharmaceutical, biomedical and agricultural chemists.
3,4,5 However, current synthetic methodologies to access these groups have not adequately met the demand, necessitating innovation. Further, harsh conditions are often required for the incorporation of fluorine on organic molecules, which many times limits late stage-synthetic applications.
6 One such group, the difluoromethylene, has been used to block metabolically labile benzylic positions,
7 or as a substitute for metabolically labile oxygen atoms.
8 Current synthetic strategies allow for several retrosynthetic disconnections; however strong oxidants,
9 bases,
10 or organometallic reagents are often required.
11 An alternate retrosynthetic disconnection could reveal a heteroatom-based nucleophile and an electrophilic
gem-difluoroakene.
12,13 In a forward reaction, a
gem-difluorinated alkene is an emerging synthon for accessing a wide variety of products, as the last several years have witnessed an explosion of synthetic functionalization reactions of
gem-difluoroalkenes.
12,13,14,15 As a result, robust and scalable procedures for converting readily available starting materials, such as aldehydes, to
gem-difluoroakene are desired.
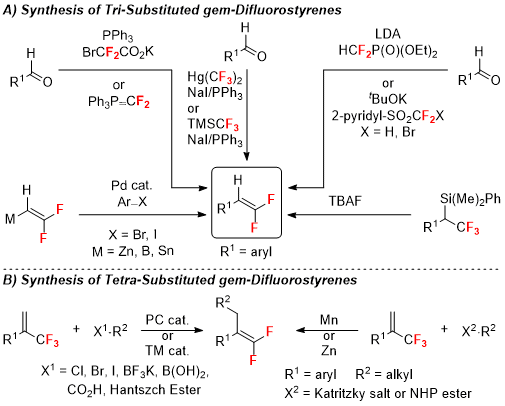
Figure 10. Strategies to access gem-difluoromethylene containing molecules
Several strategies enable access to
gem-difluoroalkenes.
14,15,16 Many of these reactions involve the conversion of aldehydes to
gem-difluoroalkenes, such as Wittig olefination reactions that exploit difluorocarbene [:CF
2] precursors, such as XCF
2CO
2K (X = Cl, Br), Ph
3P
+-CF
2CO
2- and TMS-CF
3/NaI,
16,17,18,19,20,21,22,23 or Julia-Kocienski olefination reactions using Ar-SO
2CF
2X
24,25 (Figure 10A). However, other non-olefination strategies are also well developed, such as umpolung cross-coupling of
gem-difluorovinyl nucleophiles with aryl halides (Figure 10A),
26,27,28,29 desilylative defluorination of benzyl trifluoromethanes (Figure 10A),
30 and photocatalyst/transition metal mediated defluorinative functionalization of trifluoromethylalkenes compounds (Figure 10B). Such defluorinative functionalization of trifluoromethylalkenes generates tetra-substituted alkene products, which contrasts with the tri-substituted
gem-difluorostyrenes that are synthetized herein via Wittig difluoroolefination reactions.
Beneficial aspects of the present decarboxylative Wittig olefination reactions include the broad availability of aldehyde-derived substrates, the availability and low cost of reagents (PPh
3, NaO
2CCF
2Br), the simplicity of a one-pot procedure, and the broad reaction scope that can deliver both aromatic and alkyl-derived products. In contrast, reported preparations of
gem-difluoroalkenes have several disadvantages relative to the modified Wittig-type olefination reactions reported herein (Figure 10). Other modified Wittig-type or Julia-Kocienski olefination strategies use mercury(II), which presents an unnecessary safety risk, or require cryogenic conditions, which is expensive and challenging to implement on large scales (Figure 10A).
25,31,32 Next, the desilylative defluorination of benzyl trifluoromethanes requires multiple synthetic step to prepare α-silyl-α-trifluoromethyl benzyl substrates, which involves handling of potentially explosive diazo compounds (Figure 10A). Additionally, the cross-coupling of aryl halides and vinyl nucleophiles have not yet been demonstrated with alkyl halide electrophiles, and therefore cannot deliver aliphatic
gem-difluoroalkene products (Figure 10A).
26,27,28,29 This limitation might arise from instable alkyl-palladium(II) intermediates that undergo β-H elimination processes to deliver products bearing alkenes at alternate positions. Further, photocatalytic defluorinative functionalization of vinyl-CF
3 reagents offers a mild alternative; however, the reaction has only been demonstrated on aryl-derived substrates, perhaps due to the ability of the aryl ring to stabilize a benzylic radical and benzylic anion intermediates (Figure 10B).
15 Though, transition metal-mediated defluorinative functionalization avoids the pitfalls of the photocatalytic strategy, demonstration of the method on a multi-gram scales has not been achieved (Figure 10B).
15 Finally, the reduction of Katritzky salts and NHP esters requires stoichiometric Mn or Zn, and has only been demonstrated to work on up to one gram scale (Figure 10B).
15,33The described procedure herein utilizes BrCF
2CO
2K, which is commercially available, or can be prepared on multigram scale.
22 gem-Difluoroolefination of aldehydes proceeds through a multistep mechanism initiated by the displacement of bromide on
I by PPh
3 to generate zwitterionic intermediate
II (Figure 11),
20,22 which then undergoes thermal decarboxylation to form the active ylide species
III. The nucleophilic
gem-difluorinated carbon of ylide
III then attacks the carbonyl, generating alkoxide-phosphonium zwitterion
IV. The alkoxide of
IV can then attack phosphorus to generate oxaphosphetane
V, and subsequent cycloreversion yields triphenylphosphine oxide and
gem-difluoroalkene
VI.
Figure 11. Proposed mechanism for gem-difluoroolefination of aldehydes
Practically, this preparation benefits from a controlled dropwise addition of BrCF
2CO
2K in DMF to a heated solution aldehyde and triphenylphosphine in DMF to control the rate of decarboxylation. This dropwise addition of BrCF
2CO
2K requires one less synthetic step than pre-forming zwitterion
II. An additional improvement was realized in the removal of excess triphenylphosphine, for which hydrogen peroxide is typically used as an oxidant to generate triphenylphosphine oxide that can be readily removed by filtration.
34 However, the use of superstoichiometric peroxides on multigram scales presents potential explosion hazards. To avoid such a hazard, methyl iodide was used as a less hazardous option that would react with residual PPh
3 to generate (methyl)triphenylphosphonium iodide, which is then filtered off using a short plug of silica. Additionally, the use
31P NMR enables estimation of the remaining quantity of unreacted PPh
3, which can be used to calculate the minimal amount of MeI required alkylate and remove this unreacted material. Finally, other reported preparations of
gem-difluoroalkenes typically operate below 1.0 mmol scale, which limits the build-up of a substantial amount of material for further functionalization, whereas we have used the present difluoroolefination reaction, typically on 25-50 mmol scale reproducibly for preparing a variety of substrates.
35,36,37,38Notably, it is important to distinguish this procedure from an earlier contribution by Silverstein.
39 The reported method herein uses BrCF
2CO
2K instead of ClCF
2CO
2Na, which allows the reaction to proceed at a lower temperature (90 ℃ vs. 160 ℃). In general, running a reaction at a lower temperature typically translates to improved functional group compatibility. In our hands, for this specific reaction,
gem-difluoroalkene products bearing electron-donating groups seem to decompose at higher temperatures, and the use of BrCF
2CO
2K allows the synthesis of electron-rich
gem-difluoroalkenes without significant decomposition. Additionally, in regard to reaction set-up, the use of ClCF
2CO
2Na, required a specialized heated addition funnel to fully solubilize the salt, whereas BrCF
2CO
2K is soluble in DMF at rt and does not require specialized glassware.
The described synthetic methodology has been applied to a wide variety of aldehydes to access the corresponding
gem-difluoroalkenes. For example, heterocyclic substrates including dibenzothiophenes and pyrazoles were competent substrates (Figure 12).
35,40 Further, useful functional groups including acrylates and amides chemoselectively react to generate
gem-difluoroalkene containing products. Finally, these conditions are also compatible with aliphatic aldehydes to give aliphatic
gem-difluoroalkenes.
2Figure 12. gem-Difluoroolefination tolerates a wide variety of functional groups
Overall, this Wittig
gem-difluoroolefination of aldehydes provides rapid access to
gem-difluoroalkenes on a multigram scale. Its operational simplicity and attention to safety offers an attractive synthesis of
gem-difluoroalkenes. Further, this procedure has enabled rapid access to a wide range of
gem-difluoroalkenes, which have been used in subsequent fluorine-retentive functionalization reactions with many nucleophiles.
2,12,13,14,15,35,36,37,38
Appendix
Chemical Abstracts Nomenclature (Registry Number)
4-Formyl benzonitrile; (1) (105-07-7)
PPh3: Triphenylphosphine; (603-35-0)
DMF: N,N-Dimethylformamide; (68-12-2)
4-(2,2-Difluorovinyl)benzonitrile (38936-00-4) (2)

|
Andrew John Intelli received his B.S. in Biochemistry from Stockton University, Galloway, NJ, USA in 2018, where he performed research under the guidance of Dr. Pamela Cohn and Dr. Shanthi Rajaraman. In 2019, he began his studies at the University of Kansas, Kansas before moving to Purdue University with Dr. Ryan Altman, where he is currently a sixth-year graduate student. His studies primarily focus on Pd-catalyzed C-H functionalization and fluorine-retentive functionalization of gem-difluoroalkenes. |

|
Jacob P. Sorrentino earned his B.S. in Biology with a minor in Chemistry at Nevada State College, studying under Professor Zachary Woydziak and his Ph.D. in medicinal chemistry in working under Professor Ryan Altman. Following his doctoral studies, Jacob served as a postdoctoral researcher in the laboratory of Professor Neil Garg in the Department of Chemistry and Biochemistry at the University of California, Los Angeles. Jacob now works as a medicinal chemist at Terray Therapeutics. |

|
Ryan Altman received a bachelor's degree from Creighton University, and his PhD from MIT working with Stephen Buchwald. He conducted postdoctoral studies with Professor Larry Overman at UC Irvine, and subsequently started his independent career at The University of Kansas in the Department of Medicinal Chemistry. In 2020, his group moved to Purdue to join both the Borch Department of Medicinal Chemistry and Molecular Pharmacology and the Department of Chemistry, where he is currently a Full Professor and the Steve and Lee Ann Taglienti Chair in Pharmacy. The Altman group works at the interface of synthetic organic and medicinal chemistries, with synthetic emphases in the areas of organometallic and organofluorine transformations and unique chemical reactivities enabled by fluorinated substructures. |

|
Niccolò Carlo Azzini was born in Verona, Italy. He currently attends Loughborough University in the UK and is expected to receive a BS in Chemistry in 2025. He worked for a year as an undergraduate researcher in Prof. Dirk Trauner's group at the University of Pennsylvania, focusing primarily on photopharmacology. |

|
Christoph Etling earned his B.Sc. in Chemistry from the University of Hannover, Germany, where he continued his studies in Medicinal and Natural Product Chemistry at the master's level. During this time, he joined the research group of Prof. Markus Kalesse, where he completed both his master's and doctoral theses, focusing on sesquiterpenoid total synthesis and natural product-inspired method development. In 2023, he moved to the University of Pennsylvania for a postdoctoral stay with Prof. Dirk Trauner, where he is now working in the field of photopharmacology as a Walter Benjamin Fellow (DFG). |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved