Org. Synth. 2005, 81, 121
DOI: 10.15227/orgsyn.081.0121
GENERATION AND CYCLIZATION OF 5-HEXENYLLITHIUM: 2-CYCLOPENTYLACETOPHENONE
[Ethanone, 2-cyclopentyl-1-phenyl-]
Submitted by William F. Bailey, Matthew R. Luderer, Michael J. Mealy, and Eric R. Punzalan
1.
Checked by Scott E. Denmark and Stephen L. MacNeil.
1. Procedure
Caution! tert-Butylithium is extremely pyrophoric and must not be allowed to come into contact with the atmosphere. This reagent should only be handled by individuals trained in its proper and safe use. It is recommended that transfers be carried out by using a 20-mL or smaller glass syringe filled to no more than 2/3 capacity, or by cannula. For a discussion of procedures for handling air-sensitive reagents, see Aldrich Technical Bulletin AL-134. [Note added August 2009]
A. 6-Iodo-1-hexene (1). A flame-dried, 1-L, one-necked, round-bottomed flask equipped with a magnetic stirbar and a pressure-equalizing addition funnel fitted with a nitrogen inlet adapter is charged with 10.0 g (0.100 mol) of 5-hexen-1-ol, 15.2 g (0.151 mol) of triethylamine, and 500 mL of dichloromethane (Note 1). The flask is cooled in an ice-salt bath (0 to −5°C) for 30 min and then 9.3 mL (0.12 mol) of methanesulfonyl chloride (Note 2) is added dropwise via the addition funnel. The reaction mixture is stirred at −5 to −10°C for an additional 1 hr before being transferred to a cold 1-L separatory funnel and washed successively with 150 mL of cold water, 150 mL of cold 10% (ca. 3.3N) aqueous hydrochloric acid, 150 mL of cold, saturated, aqueous sodium bicarbonate solution, and 150 mL of cold brine (Note 3). The organic layer is dried over MgSO4 and then divided into three portions (Note 4). One portion is filtered into a 300-mL, round-bottomed flask and concentrated by rotary evaporation at 8-10 mm in a 20-25°C water bath, and this is repeated with the remaining two portions to give the desired mesylate as a clear, pale-yellow oil (Note 5).
Dry acetone (200 mL) and then 18.5 g (0.12 mol) of anhydrous sodium iodide (Note 6) are added to the above 300-mL flask, which is then equipped with a magnetic stirbar and a Friedrichs condenser fitted with a nitrogen inlet adapter. The pale-yellow solution is stirred in the dark under a positive pressure of nitrogen at gentle reflux for 4 hr (Note 7). The resulting mixture is allowed to cool to room temperature and the acetone is then removed by rotary evaporation at 8-10 mm in a 20-25°C water bath. The residue is partitioned between 50 mL of pentane and 50 mL of 10% aqueous sodium thiosulfate solution by swirling the flask until all of the precipitate dissolves. The aqueous phase is discarded and the organic layer is washed with 50 mL of brine, dried over MgSO4, filtered, and concentrated by rotary evaporation at 8-10 mm in a 20-25°C water bath. The residue is then passed through a 60-mL, medium porosity, sintered-glass funnel containing ca. 20 g of alumina (Note 8) using ca. 100 mL of pentane as eluent. The pentane is removed by rotary evaporation at 8-10 mm in a 20-25°C water bath to give 17.44-17.97 g (83-86% overall from the alcohol) of 1 as a clear, colorless oil (Notes 9 and 10).
B. 2-Cyclopentylacetophenone (2). A 500-mL, two-necked, round-bottomed flask (Note 11) equipped with an egg-shaped magnetic stir bar (4 cm × 1.5 cm), argon inlet adapter, and a rubber septum is flame-dried and allowed to cool to room temperature under a positive pressure of argon. A thermometer is inserted through the septum (Note 12) and the flask is charged with 114 mL of dry, alkene-free pentane and 76 mL of anhydrous diethyl ether (Notes 13, 14). The solution is cooled to approximately −72°C using a 2-propanol-dry ice bath and 52 mL of a 1.90M solution of tert-butyllithium (t-BuLi) in heptane (98.8 mmol) is added via teflon cannula at a rate of approximately 1.7 mL/min so as to maintain an internal temperature below ca. −68°C (Notes 15, 16). A two-necked, 100-mL, round-bottomed flask equipped with a rubber septum and argon inlet adapter is flame-dried under an atmosphere of argon, allowed to cool to room temperature, and then charged with 10.0 g (47.6 mmol) of oxygen-free 6-iodo-1-hexene (1) (Note 17) and 50 mL of dry, alkene-free pentane (Note 14). The iodide solution is cooled to −72°C using a 2-propanol-dry ice bath and then transferred via a teflon cannula under a positive pressure of argon to the stirred, −72°C solution of t-BuLi at a rate of approximately 1.4 mL/min so as to maintain an internal temperature below −65°C (Note 18). The last of the iodide solution is transferred through the cannula by addition of 5 mL of dry pentane to the flask. Upon completion of the addition, the mixture is stirred at −72°C for 15 min and the cooling bath is then removed. After 3.5 hr (Note 19), the opaque, pale-yellow solution is cooled to ca. 0°C in an ice-water bath, and 4.85 g (47.0 mmol) of benzonitrile (Note 20) is added dropwise via syringe (Note 21). The cooling bath is removed, and the bright-orange solution is allowed to stir for 1 hr. The reaction mixture is then re-cooled in an ice-water bath and 35.0 mL of 10% (ca. 3.3N) aqueous hydrochloric acid is added rapidly via syringe (Note 22). Once the exotherm abates, the cooling bath is removed, and stirring is continued for 2 hr. The contents of the flask are completely transferred to a 500-mL separatory funnel by repeated washings with pentane and ether, shaken vigorously, and the aqueous layer is discarded. The organic phase is washed with 50 mL of water and 50 mL of brine, dried over MgSO4. filtered, and concentrated by rotary evaporation at 8-10 mm. Kugelrohr distillation of the residue (bath temperature ca. 180°C, 5 mm) affords 6.64-6.78 g (75-77 %) of pure 2 as a clear, colorless oil (Notes 10, 23, 24).
2. Notes
1.
5-Hexen-1-ol and triethylamine were purchased from Acros Organics and used without further purification. Alternatively,
5-hexen-1-ol may be prepared from
2-(chloromethyl)tetrahydropyran according to the literature procedure for the preparation of
4-penten-1-ol (Brooks, L. A.; Snyder, H. R.
Org. Synth. Coll. Vol. III 1955, 698).
Dichloromethane (certified ACS) was purchased from Fisher Scientific and was used as received.
2.
Methanesulfonyl chloride was purchased from Aldrich Chemical Co. and used as received. This reagent, when taken from a previously
opened container, is distilled from
phosphorus pentoxide prior to use.
3.
The separatory funnel, water,
10% hydrochloric acid, saturated
sodium bicarbonate solution, and
brine are cooled to ca. 0°C in a freezer prior to use.
4.
The dried organic layer is filtered and concentrated in ca. 150-mL portions.
5.
The mesylate is used in the next step without further purification and is essentially pure; a small quantity of residual
dichloromethane has no effect on the yield of the Finkelstein reaction. The mesylate has the following spectroscopic properties:
1H NMR (CDCl
3): δ 1.46 - 1.52 (m, 2H), 1.71 - 1.77 (m, 2H), 2.06 - 2.10 (q, J = 7.1 Hz, 2H), 2.98 (s, 3H), 4.21 (t, J = 6.6 Hz, 2H), 4.95 - 5.02 (m, 2H), 5.72 - 5.80 (m, 1H);
13C NMR (CDCl
3): δ 24.5, 28.4, 32.9, 37.2, 69.9, 115.1, 137.8.
6.
Acetone (certified ACS) was purchased from Fisher Scientific and was dried over
calcium sulfate and filtered prior to use. Anhydrous
sodium iodide was purchased from Acros Organics and used as received.
7.
Aluminum foil is wrapped around the flask and lower part of the condenser during the reaction.
8.
Alumina, adsorption, ca. 150 mesh, was purchased from Aldrich Chemical Co. and used as received.
9.
The known iodide
12 is isolated in high purity (>99 % by GC analysis) and may be used without further purification. The material displays the following properties:
nD20 = 1.5121 (lit.
3 nD20 = 1.5106); 1H NMR
pdf(CDCl
3: δ 1.46 - 1.55 (m, 2H), 1.80 - 1.87 (m, 2H), 2.05 - 2.11 (m, 2H), 3.19 (t, J = 7.0, 2H), 4.95 - 5.04 (m, 2H), 5.74-5.84 (m, 1H); 13C NMR (CDCl
3: δ 6.9, 29.6, 32.6, 32.9, 115.0, 138.1; IR (neat) 3076, 2931, 2856, 1641, 1441, 1215, 1176, 991, 912 cm
−1. The iodide may be stored indefinitely in a freezer if first saturated with dry
argon.
10.
The purity of
1 and
2 is assessed by analytical gas-liquid chromatography (GC) on a Hewlett-Packard 5890 gas chromatograph equipped with a flame-ionization detector and fitted with a 50 m × 0.2 mm HP-5 fused silica glass capillary column using linear temperature programming from an initial temperature of 150°C for 5 min to a final temperature of 200°C for 10 min at a rate of 5°C/min.
11.
A
one-necked Ace Glass 6935 flask with style-C side well (ordering code 6935-72) was used by the submitters, but the checkers found the use of a two-neck flask to be equally satisfactory.
12.
An
Omega model HH22 type J-K digital thermometer, connected to a
type K thermocouple probe inserted directly into the flask, was used to measure the temperature.
13.
Pentane (HPLC grade) was purchased from Fisher Scientific and used as received. Anhydrous
diethyl ether was purchased from Mallinckrodt Inc. and distilled under
nitrogen from a dark blue solution of
sodium and
benzophenone.
14.
The appropriate volume of solvent is most conveniently transferred under
argon from
flame-dried graduated cylinders fitted with rubber septa using
stainless steel double-tipped cannulas. Although the proportions of
pentane and
diethyl ether used as solvent are not crucial to the success of the
lithium-
iodine exchange, it is necessary to run the reaction in a solvent system that contains enough diethyl ether to render the
t-BuLi dimeric.
4 The submitters have found that a solvent mixture composed of 3:2 (by volume) of
pentane and
ether gives reproducible results.
15.
The concentration of solutions of
t-BuLi in heptane, purchased from FMC, Lithium Division, were determined immediately prior to use by titration with
2-butanol in
xylene using
1,10-phenanthroline as the indicator following the procedure described by Watson and Eastham.
5 A typical procedure is as follows: a
flame-dried, 25-mL, round-bottomed flask, equipped with a magnetic stirbar, is charged under a blanket of
argon with
3 mL of a 0.10 % (w/v) solution of 1,10-phenanthroline in dry
benzene and
0.40-0.50 mL of t-BuLi in
heptane; the violet solution is titrated with a ca. 1.0M solution of
2-butanol in xylenes until the pale yellow end point is reached. It should be noted that the nominal concentration recorded on commercial samples of an organolithium is often in error, particularly once the original seal is breached, or if the material has been stored for an extended period, and it is essential that the actual concentration be determined immediately prior to use. At least two molar equivalents of
t-BuLi must be used in the exchange reaction: a full equivalent of
t-BuLi is consumed in reaction with the
tert-butyl iodide, generated as a by-product of the exchange, to give
lithium iodide,
isobutylene, and
isobutane.
4 A slight surplus of
t-BuLi in excess of 2 molar equiv is not deleterious, since residual
t-BuLi is rapidly consumed by proton abstraction from
diethyl ether when the reaction mixture is warmed to room temperature,
4 but a large excess should be avoided.
16.
A commercial solution of
t-BuLi in
pentane may be substituted for
t-BuLi in
heptane. However, the lower volatility and higher flash-point of the
heptane solvent makes
t-BuLi in
heptane much easier and safer to handle than this alternative.
17.
The
6-iodo-1-hexene is rendered free of
oxygen by bubbling
argon through the neat iodide for at least 5 min prior to use.
18.
The exchange reaction is exothermic and too rapid an addition of the iodide will cause a temperature rise that may lead to the formation of unwanted side-products. A white precipitate of LiI is observed when approximately two-thirds of the iodide solution has been added.
19.
The solution warms slowly to room temperature; after 1.5 hr the internal temperature is approximately 15-20°C.
20.
Benzonitrile, purchased from Aldrich Chemical Co., was distilled from
calcium hydride and saturated with dry
argon for at least 5 min prior to use.
21.
An approximately 5°C exotherm was noted during the addition.
22.
The addition of the
hydrochloric acid solution results in an exotherm of approximately 20°C, accompanied by the appearance of a white precipitate and a viscous, orange oil.
23.
The known ketone
2,
6 which is isolated in high purity (>99% by GC analysis), exhibits the following physical and spectroscopic properties:
nD20 = 1.5362;
1H NMR
pdf(CDCl
3): δ 1.13 - 1.22 (m, 2H), 1.50 - 1.68 (m, 4H), 1.84 - 1.91 (m, 2H), 2.38 (7-line pattern, J = 7.7 Hz, 1H), 2.98 (d, J = 7.1 Hz, 2H), 7.43 - 7.46 (m, 2H), 7.52 - 7.56 (m, 1H), 7.94 -7.97 (m, 2H);
13C NMR (CDCl
3): δ 24.9, 32.7, 36.0, 44.7, 128.0, 128.5, 132.8, 137.2 200.31; IR (neat) 3062, 2951, 2868, 1685, 1448, 1209, 752, 690 cm
−1; Anal. calcd for C
13H
16O: C, 82.94; H, 8.57, Found: C, 82.71; H, 8.50
24.
Purification by short-path distillation (138-139°C/4.5 mm) resulted in a slightly diminished yield (
6.25 g,
71%).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The procedure described above may be used for the generation and 5-
exo cyclization of a variety of substituted 5-hexenyllithiums and, with appropriate modification, provides a general route to a variety of other olefinic
7 and acetylenic
8 organolithiums that cyclize upon warming.
9Primary alkyllithiums may be prepared rapidly and in virtually quantitative yield by treatment of a primary alkyl iodide with slightly more than two molar equiv of
t-BuLi in a hydrocarbon-ether solvent system at low temperature.
4 The exchange protocol outlined above for the preparation of
5-hexenyllithium (1) is a general one but its success often depends crucially on the appropriate choice of both halide and solvent. Primary alkyl iodides, rather than bromides, must be used to ensure that the reaction proceeds cleanly, most likely via a 10-I-2 ate-complex:
10 addition of
t-BuLi to a primary alkyl bromide often initiates radical-mediated processes. The
lithium-iodine exchange appears to involve a dimeric
t-BuLi solvate and, for this reason, the best medium for the preparation of primary alkyllithiums by
lithium-iodine exchange is a predominantly hydrocarbon solvent system that contains a quantity of a simple alkyl ether, such as
Et2O,
MTBE,
dibutyl ether, or the like, in which the
t-BuLi is predominantly dimeric. Solvent systems containing
THF,
TMEDA, or other Lewis bases that render the
t-BuLi monomeric should be avoided since elimination and coupling reactions often compete with exchange under these conditions.
Cyclization of an organolithium tethered to a suitably positioned
carbon-carbon π-bond is a thermodynamically favorable process that proceeds in a totally regioselective
exo-fashion with a high degree of stereocontrol via a transition state in which the
lithium atom is intramolecularly coordinated with the remote π-bond.
9 The stereochemical outcome of the cyclization of a substituted
5-hexenyllithium follows from the preference of the substituent to occupy a pseudoequatorial position in the chair-like transition state depicted below.
7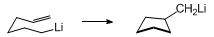
Despite the favorable thermodynamics associated with the cyclization of unsaturated organolithiums, the isomerization is often sluggish when the ring closure involves generation of a quaternary center or formation of a strained framework. In such cases it has been found that addition of lithiophilic Lewis bases such as
THF or
TMEDA facilitate the reaction.
7,9 The preparation of cuparene, a sterically congested sesquiterpene possessing two adjacent quaternary centers, illustrates the methodology.
11The predictable stereochemistry of the ring closure of substituted 5-hexenyllithiums, coupled with the ease with which the product organolithium may be functionalized, permits rational design of synthetic routes to polycyclic systems by sequential anionic cyclizations of polyolefinic alkyllithiums.
12 The preparation of stereoisomerically pure
endo-2-substituted bicyclo[2.2.1]heptanes is illustrative of this approach to polycyclic systems.
12Heterocyclic systems, such as the substituted
indoline illustrated below,
13 may also be constructed via cyclization of unsaturated organolithiums and a recent review of the preparation of
nitrogen- and
oxygen-containing heterocycles via this approach is available.
14It should also be noted that the 5-exo-trig cyclization of achiral olefinic organolithiums has been found to proceed enantioselectively when conducted in the presence of a chiral ligand that serves to render the lithium atom stereogenic. Thus, for example, (
R)-1-allyl-3-methylindoline has been prepared in 86 % ee by cyclization of an achiral aryllithium in the presence of an equivalent of
(−)-sparteine.
15
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
6-Iodo-1-hexene:
1-Hexene, 6-iodo-; (18922-04-8)
5-Hexen-1-ol; (821-41-0)
Triethylamine;
Ethanamine, N,N-diethyl-; (121-44-8)
Methanesulfonyl chloride; (124-63-0)
5-Hexen-1-ol, methanesulfonate; (64818-36-6)
Sodium iodide; (7681-82-5)
2-Cyclopentylacetophenone:
Ethanone, 2-cyclopentyl-1-phenyl-; (23033-65-0)
tert-Butyllithium;
Lithium, (1,1-dimethylethyl)-; (594-19-4)
Benzonitrile; (100-47-0)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved