Org. Synth. 2005, 82, 43
DOI: 10.15227/orgsyn.082.0043
LIPASE-CATALYZED RESOLUTION OF 4-TRIMETHYLSILYL-3-BUTYN-2-OL AND CONVERSION OF THE (R)-ENANTIOMER TO (R)-3-BUTYN-2-YL MESYLATE AND (P)-1-TRIBUTYLSTANNYL-1,2-BUTADIENE
[3-Butyn-2-ol, 4-(trimethylsilyl)-, (2R)- and Stannane, 1,2-butadienyltributyl-, (P)-]
Submitted by James A. Marshall and Harry Chobanian
1.
Checked by Peter Wipf and Joshua Pierce.
1. Procedure
A. (R)-4-Trimethylsilyl-3-butyn-2-yl Acetate (2) and (S)-4-Trimethylsilyl-3-butyn-2-yl Succinate (3). An oven dried, 1-L, round-bottomed flask, equipped with a large magnetic stir bar and rubber septum with nitrogen inlet, is charged with racemic 4-trimethylsilyl-3-butyn-2-ol (1) (10.0 g, 69.9 mmol) (Note 1) and pentane (250 mL) (Note 2). To this stirred solution is added Amano Lipase AK (Note 3) (2.00 g), freshly distilled vinyl acetate (50 mL), and pulverized, activated 4Å molecular sieves (1 g). The mixture is stirred at room temperature for 72 h at which point analysis by GC (Note 4) indicated the reaction had proceeded to 50% completion. The mixture is filtered through a medium porosity sintered glass funnel, washed with additional pentane, and concentrated via rotary evaporation affording 11.5 g of a nearly 1:1 mixture of alcohol and acetate by 1H NMR analysis. To this mixture in THF (50 mL) is added Et3N (9.1 mL, 65.0 mmol), DMAP (92 mg, 0.75 mmol) and succinic anhydride (4.15 g, 41.1 mmol) successively. The mixture is heated to reflux for 4 hr, cooled, and quenched with 40 mL of sat. aq. NaHCO3 to adjust the pH to ≥9. The solution is stirred vigorously for 1 h and diluted with ethyl acetate (EtOAc) (75 mL). The EtOAc solution is separated, washed with 10% HCl (200 mL) and brine (200 mL), dried over MgSO4, filtered, and concentrated by rotary evaporation. The resulting oil is purified by bulb-to-bulb distillation (65°C at 0.5 mmHg) (Note 5) to yield 6.10-6.11 g (94%) of (R)-4-trimethylsilyl-3-butyn-2-yl acetate (2) as a clear oil (Note 6). The aqueous phase is carefully acidified with 12 M HCl (~5 mL) to pH ~ 1 and extracted with Et2O (4 × 150 mL). The combined Et2O extracts are dried over MgSO4, filtered, and concentrated by rotary evaporation affording 8.47–8.48 g (99%) of (S)-4-trimethylsilyl-3-butyn-2-yl succinate (3) as a light yellow oil which solidified upon cooling to 0°C. The acid is carried on without further purification (Note 7).
B. (R)-4-Trimethylsilyl-3-butyn-2-ol (4). To an oven dried, 100-mL, round-bottomed flask, equipped with a magnetic stir bar and rubber septum with nitrogen inlet is added acetate 2 (6.11 g, 33.2 mmol) in 40 mL of hexanes at −78°C followed by 50 mL of DIBAL-H (1.0 M in hexanes). The solution is stirred for 10 min and poured into a rapidly stirred mixture of 300 mL of aqueous Rochelle's salt and 200 mL of Et2O. Once the Et2O layer clarifies, it is separated, dried over MgSO4, filtered, and concentrated by distillation at atmospheric pressure to yield an oil. The oil is purified by bulb-to-bulb distillation (80°C at 0.5 mmHg) to yield 4.27-4.25 g (87%) of (R)-4-trimethylsilyl-3-butyn-2-ol (4) (Note 8).
C. (S)-4-Trimethylsilyl-3-butyn-2-ol (1). To an oven dried, 250-mL, round-bottomed flask, equipped with a magnetic stir bar and rubber septum with nitrogen inlet is added a solution of the foregoing succinate 3 (8.48 g, 34.8 mmol) in 100 mL of CH2Cl2 at −78°C followed by 77 mL of DIBAL-H (1.0 M in hexanes). The solution is stirred for 10 min and the product is isolated as described for (R)-4-trimethylsilyl-3-butyn-2-ol (4). The resulting oil is purified by bulb-to-bulb distillation (75-80°C at 0.5 mmHg) to yield 4.26-4.28 g (86%) of (S)-4-trimethylsilyl-3-butyn-2-ol (1) (Notes 9 and 10).
D. (R)-4-Trimethylsilyl-3-butyn-2-yl Mesylate (5). An oven dried, 500-mL, round bottom flask equipped with a magnetic stir bar and rubber septum with nitrogen inlet is charged with (R)-4-trimethylsilyl-3-butyn-2-ol (4) (4.28 g, 30.1 mmol) and 350 mL of CH2Cl2 at −78°C. To this solution is added Et3N (8.50 mL, 61.0 mmol) followed by methanesulfonyl chloride (5.23 g, 46.0 mmol). The solution is stirred for 1 h at −78°C before being quenched with 10 mL of sat. aq. NaHCO3 solution. The solution is allowed to warm to room temperature before being concentrated at reduced pressure by rotary evaporation. The residual oil is diluted with distilled H2O (200 mL) and Et2O (100 mL). The Et2O layer is separated, dried over MgSO4, filtered, and concentrated to afford 6.07-6.09 g (91%) of (R)-4-trimethylsilyl-3-butyn-2-yl mesylate (5), which can be used without further purification (Note 11).
E. (R)-3-Butyn-2-yl Mesylate (6). To a 100-mL round-bottomed flask, equipped with a magnetic stir bar, is added a solution of (R)-4-trimethylsilyl-3-butyn-2-yI mesylate (6.09 g, 27.6 mmol) in methanol (20 mL) followed by addition of anhydrous K2CO3 (5.76 g, 42.0 mmol). After 15 min, TLC analysis (1:1; EtOAc:hexanes) of the mixture indicates complete cleavage of the TMS group. The solution is diluted with 40 mL of sat NaCl solution and extracted with diethyl ether (Et2O) (3 × 50 mL). The combined ether layers are washed with distilled H2O (3 × 50 mL) to remove residual methanol. The ether layer is separated, dried over MgSO4, filtered, and concentrated under reduced pressure (Note 12) to yield 3.48 g (85%) of (R)-3-butyn-2-yl mesylate (6) as a clear oil that can be used without further purification (Note 13).
F. (P)-(+)-3-(Tributylstannyl)-1,2-butadiene (7). An oven dried, 250-mL, round-bottomed flask equipped with a magnetic stir bar and a rubber septum with a nitrogen inlet is charged with diisopropylamine (4.0 mL, 28.0 mmol) and THF (100 mL). To this solution is added 11.2 mL (28.0 mmol) of n-BuLi (2.5 M in hexanes) at 0°C. After 30 min, tributyltin hydride (8.15 g, 28.0 mmol) (Note 14) is added dropwise and the mixture is stirred an additional 30 min. The yellow solution is then cooled to −78°C and 5.76 g (28.0 mmol) of CuBr·SMe2 is added portion-wise (Note 15). Once addition is complete, the dark solution is stirred an additional 30 min before (R)-3-butyn-2-yl mesylate (6) (Note 16) is added. After 15 min, the solution is poured into a rapidly stirred solution of 400 mL of 9:1 sat. aq. NH4Cl/NH4OH solution and 300 mL of ether. Once the ether layer clarifies, it is separated, dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting oil is purified by bulb-to-bulb distillation (120°C/0.5 mmHg) to yield 5.77-5.79 g (88%) of (P)-(+)-3-(tributylstannyl)-1,2-butadiene as a yellow oil (Note 17).
2. Notes
1.
4-Trimethylsilyl-3-butyn-2-ol was obtained from Gelest, lnc. (Submitters) or from Lancaster Research Chemicals (Checkers). All other chemicals were purchased from Aldrich Chemical Company, Inc. and used as received.
2.
The selectivity of the resolution diminished at a higher concentration of butynol. A decrease in the volume of
pentane from 250 mL to 125 mL led to acetate with er = 88:12 at 50% conversion.
3.
Amano Lipase AK from Pseudomonas fluorescens was purchased from Aldrich Chemical Company, Inc.4.
The reaction progress was monitored by GC (Carbowax, 110°C, 1°C ramp/min; acetate 5.75 min; alcohol 8.22 min. or β-DEX, 89°C, 0.2°C ramp/min, alcohol 31.30 min, acetate 32.37 min).
5.
Bulb-to-bulb (short-path) distillation was performed with an Aldrich Kugelrohr distillation apparatus.
6.
Physical characteristics of
(R)-2-acetoxy-4-trimethylsilyl-3-butyne (
2):
[α]20D +119 (c = 2.2, CHCl3); IR (film) cm
−1: 2181, 1747;
1H NMR
pdf (300 MHz, CDCl
3) δ 0.15 (s, 9H), 1.45 (d,
J = 6.7, 3H), 2.05 (s, 3H), 5.44 (q,
J = 6.7, 1H).
13C NMR (75 MHz, CDCl
3) δ −0.27, 21.1, 21.4, 60.6, 89.4, 103.5, 169.7. Analysis by GC on a β-Dex column showed a single peak at 32.0 min (80°C, 0.2°C ramp/min). The racemic acetate gave rise to peaks at 31.66 and 32.42 min under these conditions.
7.
Spectral analysis for
(S)-4-trimethylsilyl-3-butyn-2-yl succinate (
3):
1H NMR
pdf (300 MHz, CDCl
3) δ 0.16 (s, 9H), 1.47 (d,
J = 6.6, 3H), 2.50-2.80 (m, 4H), 5.49 (q,
J = 6.6, 1H).
8.
Physical characteristics of
(R)-4-trimethylsilyl-3-butyn-2-ol (
4):
[α]20D +22.4 (c = 2.01, CHCl3); IR (film) cm
−1 ν: 3334, 2175;
1H NMR
pdf (300 MHz, CDCl
3) δ 0.17 (s, 9H), 1.45 (d,
J = 6.6, 3H), 1.83 (d,
J = 5.3, 1H), 4.52 (app p,
J = 6.3, 1H);
13C NMR (75 MHz, CDCl
3) δ −0.20, 24.1, 58.5, 88.2, 107.7.
9.
(S)-4-Trimethylsilyl-3-butyn-2-ol (
1):
[α]20D −22.3 (c = 2.55, CHCl3); lit. (−25.9, c = 3.12).
210.
The acetate derivative of
(S)-4-trimethylsilyl-3-butyn-2-ol was prepared by treatment with excess
acetic anhydride,
Et3N, and
DMAP in CH2Cl2. Analysis of an aliquot by GC on a β-Dex column (80°C, 0.2°C ramp/min) revealed a 98:2 mixture of enantiomers.
11.
Physical characteristics of
(R)-4-trimethylsilyl-3-butyn-2-yl mesylate (
5):
[α]20D +98.4 (c = 2.30, CHCl3); IR (film) cm
−1: 2961, 2175;
1H NMR
pdf (300 MHz, CDCl
3) δ 0.15 (s, 9H), 1.58 (d,
J = 6.7, 3H), 3.07 (s, 3H), 5.20 (q,
J = 6.7, 1H);
13C NMR (75 MHz, CDCl
3) δ −0.63, 22.3, 38.9, 68.3, 93.4, 101.1.
12.
Care must be taken not to heat the bath during removal of the solvent from the relatively volatile mesylate solution on the
rotary evaporator.
13.
Physical characteristics of
(R)-3-butyn-2-ol mesylate (
6):
[α]20D +92.9 (c = 2.49, CHCl3); IR (film) cm
−1: 3282, 2942, 2124;
1H NMR
pdf (300 MHz, CDCl
3) δ 1.63 (d,
J = 6.6, 3H), 2.72 (d,
J = 1.8, 1H), 3.10 (s, 3H), 5.27 (dq,
J = 1.7, 6.4 Hz, 1H);
13C NMR (75 MHz, CDCI
3) δ 22.3, 39.0, 67.4, 76.3, 80.0.
14.
Tributyltin hydride was purchased from Lancaster Synthesis, Inc. With certain batches of tributyltin hydride the submitters found that extending the stirring time with LDA to 3 hours improved the yield.15.
Addition of the
CuBr·SMe2 over 5 min in small portions is important to the success of the reaction.
16.
After addition of the mesylate to the stirred reaction mixture, it is necessary to quench the reaction within 10 min to minimize
copper-catalyzed racemization of the allenylstannane. In our original communication of this methodology
3 we noted that, contrary to the findings of others
4, who reported partial racemization of allenes prepared through additions of alkylcuprates to propargylic esters, the acetoxymethyl stannane (eq, R
1 = AcOCH
2, R
2 = Me) was not racemized by prolonged exposure to the stannylcupration conditions. It appears that, perhaps not surprisingly, structural factors can influence the configurational stability of allenes prepared in this manner. It is also possible that contaminants in the copper salts or impurities of an undetermined nature could be responsible for the racemization, which is mechanistically obscure at this time.
17.
Physical characteristics of
(P)-(+)-3-(tributylstannyl)-1,2-butadiene (
7):
[α]20D +61.2 (c = 3.31, CHCl3); IR (film) cm
−1: 2924, 1928;
1H NMR
pdf (300 MHz, CDCl
3) δ 0.80-1.70 (m, 30 H), 4.45-4.71 (m, 1H), 4.87-5.12 (m, 1H).
13C NMR (75 MHz, CDCl
3) δ 10.3, 13.7, 27.2, 28.9, 74.3, 75.2, 209.0. The ee of the stannane was determined by analysis of the product obtained through
BF3·OEt2-promoted addition to
isobutyraldehyde. To a stirred solution of
(P)-(+)-3-(tributylstannyl)-1,2-butadiene (1.97 g, 57.5 mmol) and
isobutyraldehyde (300 mg, 42 mmol) in
CH2Cl2 at −78°C was added
BF3·OEt2 (2.18 mL, 172 mmol). The solution was maintained at −78°C for 1 hr then quenched by the addition of
sat. aq. NaHCO3 and warmed to rt. The
CH2Cl2 layer was removed and stirred vigorously with KF-on-Celite
5 and
MgSO4. After 1 hr, analysis of an aliquot by GC on a β-Dex column indicated a 98:2 ratio of enantiomers, and a >99:1 ratio of diastereomers (80°C, 0.2°C ramp/min; alcohol 21.8 min).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The present resolution procedure is based on a report by Burgess and Jennings,
2 but with several key modifications.
- To minimize losses of the volatile products during solvent removal, pentane was substituted for hexane.
- For the same reason, and to facilitate larger scale resolutions, the substrate concentration was increased from 0.1 to ~0.2 M. However, an increase to 0.4 M led to diminished efficiency.
- In the original report, the (S)-alcohol 1 and (R)-acetate 2 were separated by column chromatography which led to isolated yields of 90% for the acetate and only 54% for the alcohol. In the present procedure the alcohol is separated from the acetate by conversion to the half succinate, which is extracted with aqueous sodium bicarbonate. Purification of the acetate is achieved by short-path distillation. After cleavage of the ester groupings through reduction with DIBAL-H, both the (S) and (R)-alcohols are isolated following distillation in ~86% overall yield.
The
(R)-TMS butynyl mesylate 5, or its enantiomer, are converted
in situ to the
(P)-allenyl indium or zinc reagents by reaction with
InI or
Et2Zn utilizing
Pd(OAc)2·PPh3 as the precatalyst.
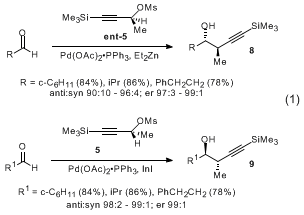
6 These metallations are conducted in the presence of various aldehydes to produce
anti homopropargylic alcohol adducts
8 or
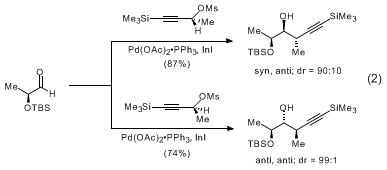
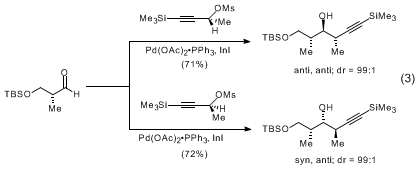
9 of high diastereomeric and enantiomeric purity (eq 1). Additions to chiral α and β-oxygenated aldehydes proceed with a high degree of reagent control to yield
syn,
anti and
anti,
anti stereotriads (eq 2, 3).
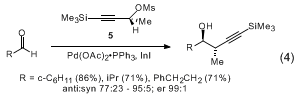
7 These additions can also be effected with the corresponding allenyl
indium and
zinc reagents derived from the terminal alkynyl mesylate
6, or its enantiomer (eq 4).
8 However, those additions proceed with significantly lower diastereoselectivity, most notably with unbranched aldehydes.
Propargylic mesylates, such as
6, can be converted to (
P)-allenylstannanes through S
N2' displacement of the mesylate with a
tributyltin cuprate reagent (eq 5).
9,10 These reagents are formed with inversion of stereochemistry and most are sufficiently stable to be purified by distillation. The reaction proceeds with high levels of enantioselectivity, provided the product is removed from the cuprate salts within 10 min. The allenyl
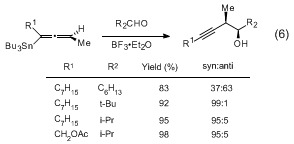
tin reagents afford
syn homopropargylic alcohol adducts upon addition to aldehydes in the presence of excess
BF3·OEt2 (eq 6). Matching/mismatching occurs in additions to α-chiral aldehydes (eq 7). Allenylstannanes undergo transmetallation with
BuSnCl3,
SnCl4, or
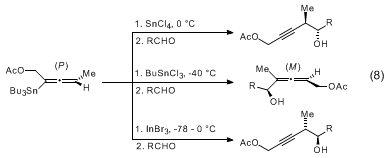
InX3 salts in the presence of aldehydes to afford allenylcarbinols or
anti homopropargylic alcohols (eq 8).
11,12,13 The
SnCl4 transmetallations proceed with overall inversion of stereochemistry whereas the
InX3 reactions lead to allenylindium intermediates with retention of stereochemistry (eq 9).
14 The resulting allenyl
SnCl3 and
InX2 reagents afford enantiomeric
anti homopropargylic alcohols upon addition to aldehydes (eq 8). The
BuSnCl3 reaction proceeds via the transmetallated propargylic stannanes (eq 10). All three of the foregoing additions proceed through cyclic transition states.
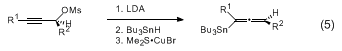
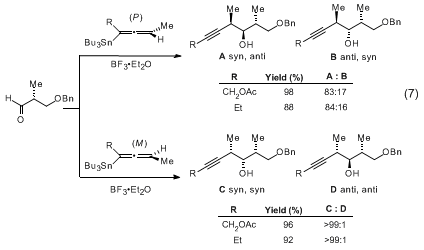
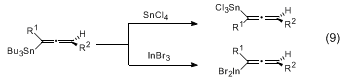
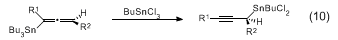
Additions to α-chiral aldehydes exhibit matching/mismatching characteristics (eq 11-13). Through appropriate selection of chiral partners and allenylmetal reagents, any of the eight possible stereotriads can be prepared by this methodology. The allenylcarbinol products are stereospecifically converted to dihydrofurans upon treatment with
5-10 mol% AgNO3.
13Chiral allenylmetal reagents have been utilized with excellent effectiveness in the synthesis of a variety of polyketide natural products of marine origin.
15,16 An attractive feature of the methodology is the ability to incorporate the alkyne function of the adducts as a reactive component for elaboration of the polypropionate and polyacetate backbone of these compounds.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(R)-4-Trimethylsilyl-3-butyn-2-yl acetate:
3-Butyn-2-ol, 4-(trimethyl silyl)-,acetate; (129571-78-4)
(S)-4-Trimethylsilyl-3-butyn-2-yl succinate:
Butanedioic acid, mono [(1S)-1-methyl-3-(trimethylsilyl)-2-propynyl] ester; (375395-73-6)
4-Trimethylsilyl-3-butyn-2-ol:
3-Butyn-2-ol, 4-(trimethylsilyl)-; (6999-19-5); (2R)-(121522-26-7); (2S)-(12155-27-8)
(R)-4-Trimethylsilyl-3-butyn-2-yl mesylate:
3-Butyn-2-ol, 4-(trimethylsilyl)-, methanesulfonate, (2R)-; (200440-90-0)
(R)-3-Butyn-2-yl mesylate:
3-Butyn-2-ol, methanesulfonate, (2R)-; (121882-95-4)
Vinyl acetate:
Acetic acid ethenyl ester; (1008-05-4)
DIBAL-H (Diisobutylaluminum hydride):
Aluminum, hydrobis (2-methylpropyl)-; (1191-15-7)
Triethylamine:
Ethanamine, N,N-diethyl-; (121-44-8)
Mesyl chloride:
Methanesulfonyl chloride; (124-63-0)
Butyllithium:
Lithium, butyl-; (109-72-8)
Tributyltin hydride:
Stannane, tributyl-; (688-73-3)
Cuprous bromide-dimethylsulfide complex:
Copper, [thiobis(methane)]; (54678-23-8)
(P)-1-Tributylstannyl-1,2-butadiene:
Stannane, 1,2-butadienyltributyl-; (202119-26-4)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved