Org. Synth. 2006, 83, 61
DOI: 10.15227/orgsyn.083.0061
SYNTHESIS OF 2α-BENZYLOXY-8-OXABICYCLO[3.2.1]OCT-6-EN-3-ONE BY [4 + 3] CYCLOADDITION
Submitted by María Vidal-Pascual, Carolina Martínez-Lamenca, and H. M. R. Hoffmann
1.
Checked by Timothy E. Long and Marvin J. Miller.
1. Procedure
A. 1,1-Bis(benzyloxy)propan-2-one (2). A one-necked, 100-mL, round-bottomed flask equipped with a magnetic stirring bar is charged with pyruvic aldehyde dimethyl acetal (12.1 mL, 100 mmol) in cyclohexane (50 mL), benzyl alcohol (22.8 mL, 220 mmol) and p-toluenesulfonic acid monohydrate (0.95 g, 5 mmol) (Note 1). The resulting mixture is heated at reflux for 2 h using a Dean–Stark separator for the removal of methanol. When the reaction is complete (approximately 2 h), approximately 8.1 mL (200 mmol) of MeOH is obtained. The reaction mixture is cooled to room temperature and washed with saturated potassium carbonate solution (25 mL) and water (20 mL). The aqueous layer is extracted twice with cyclohexane (2 × 50 mL). The combined organic phase is dried (Na2SO4), filtered, evaporated and the crude black oil is purified by column chromatography using a 10-cm diameter column packed with 900 g silica gel (Note 2) and eluting with 2.1 L of MTBE/cyclohexane (1:20) to afford keto acetal 2 as a yellowish oil (22.3 g, 83%) (Note 3).
B. [1,1-(Bis-benzyloxymethyl)vinyloxy]triethylsilane (3) (Note 4). An LDA solution is prepared in a 250-mL, two-necked, round-bottomed flask equipped with a nitrogen inlet and nitrogen balloon, magnetic stirring bar and rubber septum by adding BuLi (1.6 M solution in hexane, 22.5 mL, 36 mmol) (Note 5) into a solution containing diisopropylamine (5.1 mL, 36 mmol) (Note 6) in THF (36 mL) (Note 7) at −78 °C (Note 8). The resulting mixture is stirred for an additional 15 min at room temperature. A separate two-necked, 250-mL round-bottomed flask is equipped with two septa and a nitrogen balloon. Dibenzyl acetal 2 (8.1 g, 30 mmol) and chloro-triethylsilane (7.5 mL, 45 mmol) (Note 9) are dissolved in THF (30 mL) under a nitrogen atmosphere. This mixture is cooled to −78 °C and the LDA solution is added by cannula over the course of 10 min. Triethylamine (18.8 mL, 135 mmol) (Note 10) was immediately added by syringe over the course of 10 min. The resulting reaction mixture is stirred for 16 h at −78 °C. Water (25 mL) is added, the cooling bath is removed, and the mixture is stirred until it reaches room temperature. The aqueous phase is extracted twice with cyclohexane. After being dried over sodium sulfate (Na2SO4) the organic solution is concentrated under vacuum and purified by column chromatography (6-cm diameter column, 360 g silica gel, MTBE/cyclohexane (1:100, 9 L) with 0.1% Et3N) (Note 11) to give 3 as a yellow oil (8.9 g, 77%) (Notes 12, 13).
C. 2α-Benzyloxy-8-oxabicyclo[3.2.1]oct-6-en-3-one (4) A 250-mL, one-necked, round-bottomed flask capped with a rubber septum and equipped with a nitrogen balloon and magnetic stirring bar is charged under nitrogen atmosphere with silyl enol ether 3 (9.2 g, 24 mmol) (Note 14) and dichloromethane (25 mL) (Note 15). The mixture is cooled to −78 °C, and furan (1.8 mL, 24 mmol) (Note 16) is added by syringe. After 15 min at −78 °C, TMSOTf (0.46 mL, 2.4 mmol) is added by syringe (Note 17). The mixture is stirred for 30 min at −78 °C and then a saturated solution of sodium bicarbonate (25 mL) is added. The cooling bath is removed and the flask is shaken thoroughly until the mixture reaches room temperature. The aqueous layer is extracted with dichloromethane (3 × 25 mL) and the combined organic phase is dried (Na2SO4). After removal of the solvent under reduced pressure, the crude product is purified by column chromatography (5-cm diameter column, 250 g silica gel, MTBE/cyclohexane (1:3)) giving cycloadduct 4 as a colorless oil (3.72 g, 70%) (Note 18). On storage in the freezer at −25 °C, the product crystallizes from tert-butyl methyl ether to give a white solid (2.87 g, 54%) mp 64–65 °C (Notes 19-20).
2. Notes
1.
Pyruvic aldehyde dimethyl acetal was purchased from Acros Organics.
Benzyl alcohol was purchased from Lancaster.
p-Toluenesulfonic acid was purchased from Aldrich.
2.
Silica gel (230-400 mesh) was obtained from Macherey Nagel.
3.
Spectral data for
1,1-bis(benzyloxy)propan-2-one 2:
1H NMR
pdf (500 MHz, CDCl
3) δ: 2.33 (s, 3 H), 4.68 (d,
J = 12 Hz, 2 H), 4.78 (d,
J = 12 Hz, 2 H), 4.84 (s, 1 H), 7.41–7.45 (m, 10 H);
13C NMR (75 MHz, CDCl
3) δ: 25.0, 69.2, 101.0, 127.4, 128.0, 128.5, 137.0, 203.7. EI-MS (
m/z): 271 (5), 259 (15), 228 (10), 182 (18), 181 (100), 165 (14); HRMS (FAB) (
m/z): calcd for C
17H
19O
2 (M
+ + 1) 271.1334, observed 271.1308.
4.
The preparation of
[(1,1-bis(benzyloxy)methyl)vinyloxy]-triethylsilane (
3) is appropriate for scales of
30 mmol of dibenzyl acetal or less. See
Note 20 for information regarding larger scale reactions.
5.
A commercial solution of
1.6 M butyllithium in hexane from Acros Organics was used.
6.
Diisopropylamine was purchased from Acros and was distilled from
KOH pellets and stored over solid KOH, purchased from Fisher Scientific.
7.
Tetrahydrofuran supplied by Mallinckrodt is dried by distillation from
sodium and
benzophenone under an argon atmosphere.
8.
The reaction mixture is cooled to −78 °C using dry
ice-acetone bath or a cryostat.
9.
Chlorotriethylsilane from both Acros Organics and Gelest, Inc. was used.
10.
Triethylamine was dried and distilled from and stored over KOH pellets purchased from Fisher Scientific.
11.
The checkers found that addition of
triethylamine to the eluent is required to prevent decomposition of the silyl enol ether during chromatography.
12.
Spectral data for
[(1,1-bis(benzyloxy)methyl)-vinyloxy]triethylsilane 3:
1H NMR
pdf (300 MHz, CDCl
3) δ: 0.69 (q,
J = 7.8 Hz, 6 H), 0.95 (t,
J = 8.1 Hz, 9 H), 4.37 (d,
J = 1.2 Hz, 1 H), 4.57 (d,
J = 12 Hz, 2 H); 4.63 (d,
J = 11.7 Hz, 2 H), 4.70 (s, 1 H), 4.89 (s, 1 H), 7.31–7.26 (m, 10 H);
13C NMR (125 MHz, CDCl
3) δ: 4.9, 6.7, 67.6, 92.3, 99.2, 127.5, 127.8, 128.4, 138.2, 153.8. EI-MS (
m/z): no M
+, 279 (9), 249 (4), 248 (3), 193 (4), 187 (6), 181 (7), 159 (14), 157 (13), 115 (17), 91 (100).
13.
The submitters report that this procedure can also be used for the preparation of
3,3-bis-benzyloxy-2-trimethylsilyloxypropene by using
chlorotrimethylsilane (from Acros Organics) instead of
chlorotriethylsilane. In that case, the resultant
trimethylsilyl enol ether cannot be purified by column chromatography due to its acid sensitivity, so the crude product is used directly in the [4 + 3] cycloaddition reaction
(Note 20).
14.
An alternative to
silyl enol ether 3 as oxyallylic cation precursor is
3,3-bis-benzyloxy-2-trimethylsilyloxypropene (Notes
13,
20).
15.
Commercial
dichloromethane was freshly distilled from
CaH2.
16.
Commercial
furan (99%, stabilized with BHT) from Acros Organics was used without further purification.
2,5-Dimethylfuran (99%) from Acros Organics was used by the submitters in the unchecked [4 + 3] cycloaddition (85% yield of cycloadduct) following the same procedure as for
furan.
17.
Commercial
trimethylsilyl trifluoromethanesulfonate (99%) from Acros Organics was used.
Ten mole % (0.1 equiv) of TMSOTf was sufficient to catalyze the [4 + 3] cycloaddition.
18.
The checkers observed that the oil partially solidifies to a white solid upon standing. The submitters report that residual
benzyl alcohol can be removed by heating the oil at 100 °C for two hours using a Kugelrohr apparatus. The checkers did not identify the presence of
benzyl alcohol by NMR analysis and did not perform the heating.
19.
Spectral data for
2α-benzyloxy-8-oxabicyclo[3.2.1]oct-6-en-3-one 4:
1H NMR
pdf (500 MHz, CDCl
3) δ: 2.37 (d,
J = 15.5 Hz, 1 H), 2.75 (dd,
J = 15, 5 Hz, 1 H), 4.13 (d,
J = 5 Hz, 1 H), 4.64 (d,
J = 12 Hz, 1 H), 4.91 (dd,
J = 5, 1.5 Hz. 1 H), 4.99 (m, 2 H), 6.30 (dd,
J = 6.5, 1.5 Hz, 1 H), 6.34 (dd,
J = 6, 2 Hz, 1 H), 7.39–7.31 (m, 5 H);
13C NMR (125 MHz, CDCl
3) δ: 46.1, 73.7, 78.6, 80.0, 84.3, 128.1, 128.7, 132.0, 134.8, 137.8, 205.2. IR (CHCl
3) cm
−1: 1724 (very strong), 1112 (very strong), 731 (strong), 697 (strong); EI-MS (rt): 230 (6, M
+), 201 (4), 158 (38), 139 (31, M
+-Bn), 121 (10), 108 (25), 91 (100), 81 (30), 77 (14), 69 (23). Anal. Calcd for C
14H
14O
3 : C, 72.89; H, 6.05. Found: C, 73.03; H, 6.13.
20.
Alternatively, the [4 + 3] cycloadduct has been prepared by the submitters using inexpensive
chlorotrimethylsilane (Note 13) as follows:
A two-necked, 250-mL, round-bottomed flask equipped with a magnetic stirring bar is charged with dibenzyl acetal 2 (27 g, 100 mmol) in anhydrous DMF (33 mL) (Dimethylformamide (DMF) is dried over powdered BaO for 2 weeks, followed by decanting and then distilling under reduced pressure.) and chlorotrimethylsilane (28.6 mL, 225 mmol) (Note 13) and heated at 75 °C. Triethylamine (38.9 mL, 280 mmol) (Note 10) is added slowly by syringe pump (60 mL/h). The reaction mixture is refluxed for 16 h at 75 °C and the reaction mixture becomes thicker and dark brown. The reaction mixture is cooled to 0 °C, washed with 30 mL of a cold saturated solution of ammonium chloride (30 mL) and water (approximately 15 mL). The aqueous phase is extracted with cyclohexane (4 × 50 mL). The combined organic phase is dried (Na2SO4) and the solvent removed with a rotatory evaporator. The resulting brown viscous oil is used directly in the [4 + 3] cycloaddition as described in procedure C.
When the 3,3-bis-benzyloxy-2-trimethylsilyloxypropene is used as the oxyallylic cation precursor, the yield of the [4 + 3] cycloadduct with furan is 41 % (two steps). The yield of the [4 + 3] cycloadduct with 2,5-dimethylfuran is 53 % (two steps). The procedure has been used in a reaction using 130-mmol of starting dibenzyl acetal.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
In view of the wide synthetic interest of [4 + 3] cycloadducts of oxyallyl cations and dienes, a variety of methods have been developed for the generation and capture of appropriate oxyallyl cations. The classical routes to generate these reactive intermediates start from halogenated precursors:
2 α,α'-Dihalo ketones or α-monohalo ketones. For example,
2,4-dimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one (5α,α) has been prepared from both a dihalo-
3a and a monohaloketone
3b and has served as a
meso-configured workhorse for exploring desymmetrization of the seven-carbon backbone with its three pro-stereogenic sp
2 centers. Allylic halides and silver salts have been used also.
2a,4 Further, the use of trialkylsilyloxyallyl cations from
1,1-dimethoxyacetone in [4 + 3] cycloadditions with furans has been reported.
5In the procedure described here, a triethylsilyloxyallylic cation with a π-donating benzyloxy substituent at the allylic terminus is generated via treatment of
triethylsilyl enol ether 3 with
TMSOTf in catalytic amount and, in presence of the 4π-component, the [4 + 3] cycloaddition proceeds smoothly giving 2α-benzyloxy-8-oxabicyclo[3.2.1]oct-6-en-3-ones. If
furan is replaced by
2,5-dimethylfuran, then cycloaddition yields are higher (leading to
6). Increasing the equivalents of diene does not alter the yield appreciably. The availability of oxabicycle
6 opens a short route to the dioxatricyclic core of the marine metabolite dictyoxetane (
6 →
7)
6a and to functionalized scopolines.
6b An asymmetric variant of the [4 + 3] cycloaddition involving chiral allyl cations has also been accomplished affording
8 with high
ee.
7Oxabicycle
8 is a useful building block and is endowed with all the chiral information for the
de novo construction of C-glycosides. For example, axial hydroxylation (
8 →
9) proceeds smoothly by treatment of the derived TES enol ether with
m-chloroperbenzoic acid provided that
wet THF is used as solvent.
8 The use of dilute solutions of
dimethyldioxirane as oxidant which is toxic and dangerous, is obviated.
9 Epimerization of
9 affords the diequatorial epimer
10. Starting from
8 and parent
8-oxabicyclo[3.2.1]oct-6-en-3-one, a complete series of C-glycosides with fully resolved seven-carbon backbone polyol stereochemistry and with complete anomeric control has been prepared (Scheme 1).
8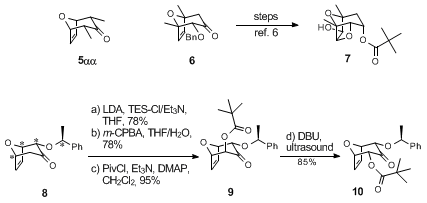
8-Oxabicyclo[3.2.1]oct-6-en-3-ones have been converted into [3.3.1]lactone acetals, which are versatile synthetic intermediates and allow further stereocontrolled C-C, C-S, C-P
+Ph
3X
− bond formation. C-C Bonds and extended carbon chains are formed with complete anomeric control in the presence of a Lewis acid (
Me3SiOTf) and silylated nucleophile in acetonitrile (Scheme 2).
10 8-Oxabicyclo[3.2.1]oct-6-en-3-ones have been used as key precursors in natural product syntheses, as accomplished by the total synthesis of hinokitiol
11 and callistatin A.
12Intramolecular [4 + 3] cycloadditions have also been studied.
13 Recently, metathesis of substituted oxabicyclics combined with spiroannulation has been reported.
14 Ru-Catalyzed asymmetric ring-opening/cross-metathesis provides another efficient enantioselective route to functionalized tetrahydropyrans.
15 Vinylic nonaflates from 8-oxabicyclo [3.2.1] oct-6-en-3-ones have been used to elaborate the bicyclic scaffold.
16
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
1,1-Dimethoxy-2-propanone:
α,α-Dimethoxyacetone; (6342-56-9)
Chlorotriethylsilane:
Silane, chlorotriethyl-; (994-30-9)
Trimethylsilyl trifluoromethanesulfonate:
Trifluoromethanesulfonic acid trimethylsilyl ester;
Trimethylsilyl triflate; (27607-77-8)
Chlorotrimethylsilane:
Silane, chlorotrimethyl-; (75-77-4)
2,5-Dimethylfuran; (625-86-5)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved