Org. Synth. 2010, 87, 362
DOI: 10.15227/orgsyn.087.0362
SYNTHESIS OF A N-MESITYL SUBSTITUTED AMINOINDANOL-DERIVED TRIAZOLIUM SALT
[(5aS,10bR)-5a,10b-Dihydro-2-(2,4,6-trimethylphenyl)-4H,6H-indeno[2,1-b]-1,2,4-triazolo[4,3-d]-1,4-oxazinium chloride]
Submitted by Justin R. Struble and Jeffrey W. Bode
1.
Checked by Yajing Lian and Huw M. L. Davies.
1. Procedure
A. 2-Mesitylhydrazinium chloride (1). A 3-necked, 1-L round-bottomed flask is equipped with a Teflon-coated overhead stirrer in the center port (Note 1) and placed in a −10 °C acetone bath (Note 2). The flask is charged with conc. HCl (26.5 mL) and water (55
mL) (Notes 3 and 4). Another port of the reaction vessel is equipped with a 60-mL pressure-equalizing addition funnel and charged with 2,4,6-trimethylaniline (15.0 mL, 107 mmol, 1.0 equiv) (Note 5), which is added dropwise over a period of approximately 5 min resulting in a thick white slurry. Stirring is continued for an additional 15 min (Notes 6 and 7). At this time, the third port is equipped with a thermometer to monitor the internal temperature of the reaction. The addition funnel is replaced with a new 60-mL addition funnel that is charged with a freshly prepared solution of NaNO2 (7.38 g, 107 mmol, 1.00 equiv) in water
(20 mL) (Note 4). This solution is added dropwise so as to maintain an internal temperature of < 5 °C (Note 8). The addition funnel is washed with H2O
(5 mL) (Note 4) and stirring is continued for an additional 30 min. The addition funnel is replaced with a new addition funnel which is charged with a solution of SnCl2•2H2O (60.4 g, 268 mmol, 2.50 equiv) in 1:1 conc. HCl/H2O
(60 mL) (Note 9). The SnCl2solution is added dropwise over 4 h maintaining an internal reaction temperature of < 5 °C. The addition funnel and thermometer are removed. The heterogeneous orange mixture is stirred at 0 °C for 1 h before being allowed to warm to ambient temperature and vigorous stirring is maintained for 16 h. The mixture is cooled by placing the reaction vessel in the refrigerator operating at 4 °C for 2 h. The orange precipitate is collected by suction filtration (Note 10) and the reaction vessel is washed with brine (5 × 50 mL) that is poured over the collected solid with each wash. The orange solid is transferred to a 1-L round-bottom flask and Et2O
(300 mL) (Note 11) and a Teflon-coated magnetic stir bar are added. The heterogeneous mixture is placed in an ice/brine bath and the flask is equipped with a pressure-equalizing addition funnel charged with aq 10 M NaOH
(200 mL) that is added over approximately 30 min. An additional 100 mL of H2O (Note 4) is added and stirring is continued for 1 h before the biphasic mixture is allowed to warm to ambient temperature and transferred to a 1-L separatory funnel. The organic layer is separated and the aqueous phase extracted with Et2O (2 × 200 mL). The organic fractions are combined and washed with brine
(250 mL), dried over MgSO4, and filtered (Note 10) into a dry 1-L, two-necked, round-bottomed flask equipped with an overhead stirrer. The other neck is filled with a rubber septum. The drying agent is washed with Et2O (3 × 30 mL). The orange solution is placed under an atmosphere of N2(g) and immersed in an ice/brine bath. A solution of HCl (4 M in dioxane, 27.0 mL, 108 mmol, 1.0 equiv) is added via syringe over 15 min inducing a white precipitate. Stirring is maintained for 30 min and the solution is allowed to warm to ambient temperature. The overhead stirrer is removed and washed with Et2O
(20 mL). The solvent is evaporated under reduced pressure and MeOH
(400 mL) is added. The heterogeneous mixture is heated to 50 °C until the solid is dissolved [approximately for 10 min] and the solvent is then removed to dryness using a rotary evaporator under vacuum to afford a crystalline product (Note 12). Et2O
(500 mL) is added and the center neck is equipped with a water condenser, while the other neck has a glass stopper. The reaction mixture is heated to reflux for 30 min and then the heterogeneous mixture is cooled to room temperature. The pale orange precipitate is collected by suction filtration (Note 10) and washed with Et2O
(50 mL). The crude solid is transferred to a 200-mL, round-bottomed flask and the residual volatiles are removed under high vacuum (ambient temperature, <0.75 mmHg, 12 h). A solution of 200 proof EtOH/Et2O (5:1, 60 mL) is added, and the suspension is placed into a sonicating bath for 30 min (Note 13). The pale orange solid is collected by suction filtration (Note 11) and washed with a solution of 200 proof EtOH/Et2O (1:1, 3 × 30 mL). The filtrate is concentrated using a rotary evaporator (35 °C, 35 mmHg) to produce an orange crystalline solid. A solution of 200 proof EtOH/Et2O (1:1, 15 mL) is added, and the suspension is placed into a sonicating bath for 5 min [Note 13]. The pale orange solid is collected by suction filtration (Note 10), and washed with a solution of 200 proof EtOH/Et2O (1:1, 3 × 5 mL). The collected solids are combined and transferred to a 200-mL, round-bottomed flask and the residual volatiles are removed under high vacuum (ambient temperature, <0.75 mmHg, 12 h) to afford 7.20-7.92 g (36-40% yield) of 2-mesitylhydrazinium chloride (1) as a pale, orange powder (Note 14).
B. (4aR,9aS)-3-Methoxy-2,4a,9,9a-tetrahydroindeno [2,1-b][1,4]oxa-zine (2). A flame-dried, 500-mL, round-bottomed flask is equipped with a Teflon-coated stir bar (Note 15), (4aR,9aS)-4,4a,9,9a-tetrahydroindeno[2,1-b][1,4]oxazin-3(2H)-one (7.00 g, 37.0 mmol, 1.00 equiv) (Note 16), CH2Cl2
(185 mL) (Note 17) and trimethyloxonium tetrafluoroborate (6.57 g, 44.4 mmol, 1.20 equiv) (Note 18). The flask is equipped with a septum, placed under an atmosphere of argon through a needle inserted into the septum and stirred at ambient temperature for 16 h. The light tan solution is immersed in an ice/water bath and aq sat NaHCO3 (150) is added over a period of 1.5 h. Stirring is maintained for an additional 1 h, then the biphasic solution is transferred to a separatory funnel, the organic phase separated, and the aqueous phase extracted with CH2Cl2(4 × 100 mL). The combined organic fractions are dried over MgSO4, filtered, and concentrated using a rotary evaporator (25 °C, 75 mmHg). The residual volatiles are removed under high vacuum (ambient temperature, <0.75 mmHg, 12 h) to afford 6.89-7.03 g (92-93% yield) of (4aR,9aS)-3-methoxy-2,4a,9,9a-tetrahydroindeno [2,1-b][1,4]oxazine (2) as a pale brown solid which is used without further purification (Note 19).
C. (Z)-2-Mesityl-1-((4aR,9aS)-4,4a,9,9a-tetrahydroindeno[2,1b][1,4] oxazin-3(2H)-ylidene)hydrazinium chloride (3). A flame-dried, 500-mL round-bottomed flask is charged with a Teflon-coated stir bar (Note 15), 2-mesitylhydrazinium chloride (1) (6.44 g, 34.4 mmol, 1.00 equiv) and MeOH
(138 mL) (Note 20), resulting in a light orange solution after stirring at ambient temperature for 5 min. To this solution is added (4aR,9aS)-3-methoxy-2,4a,9,9a-tetrahydroindeno [2,1-b][1,4]oxazine (2) (7.00 g, 34.4 mmol, 1.00 equiv) in a single portion and the mixture is stirred at ambient temperature until a red homogeneous solution forms (ca. 5 min). A catalytic amount of anhydrous HCl (4 M in dioxane, 0.86 mL, 0.10 mmol) is added to the solution. The reaction flask is equipped with a water-jacketed reflux condenser and immersed in a 60 °C silicon oil bath and the reaction mixture is stirred under an atmosphere of N2(g) for 48 h. The mixture is allowed to cool to ambient temperature and the volatiles are removed using a rotary evaporator (35 °C, 30 mmHg) followed by high vacuum (ambient temperature, <0.75 mmHg) to afford a crude orange solid. The crude material is suspended in EtOAc
(125 mL) (Note 21), a Teflon-coated magnetic stir bar (Note 15) is added, and the flask equipped with a water-jacketed reflux condenser. The mixture is stirred vigorously in a silicon oil bath at 90 °C under an atmosphere of N2(g) for 30 min, causing a light yellow precipitate to form. The flask is removed from the oil bath, allowed to cool to ambient temperature with vigorous stirring and immersed in an ice/water bath at 0 °C with vigorous stirring. The precipitate is collected by suction filtration and washed with EtOAc (3 × 20 mL). The light yellow solid is transferred to a 20 mL vial and residual volatiles are removed under high vacuum (ambient temperature, <1 mbar, 12 h), affording 9.96-10.1 g (81-82 %) of (Z)-2-mesityl-1-((4aR,9aS)-4,4a,9,9a-tetrahydroindeno-[2,1b][1,4]-oxazin-3-(2H)ylidene)hydrazinium chloride (3) as a light yellow powder (Notes 22 and 23).
D. (5aS,10bR)-5a,10b-Dihydro-2-(2,4,6-trimethylphenyl)-4H,6H-indeno[2,1-b]-1,2,4-triazolo[4,3-d]-1,4-oxazinium chloride (4). An oven-dried, 350-mL sealed tube is charged with a Teflon-coated magnetic stir bar (Note 15), (Z)-2-mesityl-1-((4aR,9aS)-4,4a,9,9a-tetrahydroindeno-[2,1b][1,4]oxazin-3(2H)-ylidene)hydrazinium chloride (3) (9.00 g, 25.1 mmol, 1.00 equiv), chlorobenzene (25.5 mL) (Note 24), triethylorthoformate (33.4 mL, 201 mmol, 8.0 equiv) (Note 25) and anhydrous HCl (4 M in dioxane, 6.28 mL, 25.1 mmol, 1.00 equiv) (Note 26). The vessel is purged with N2(g), sealed, and immersed in a silicon oil bath at 120 °C. The tan heterogeneous mixture is stirred for 1 h, resulting in a brown homogeneous solution. The solution is allowed to cool to ambient temperature, transferred to a 250-mL, round-bottomed flask (Note 27), and the solvent is removed on a rotary evaporator (60 °C, 15 mmHg) followed by high vacuum (ambient temperature, <0.75 mmHg, 1.5 h), affording a brown solid. A Teflon-coated stir bar and toluene
(80 mL) is added to the flask, which is equipped with a water-jacketed reflux condenser and immersed in a 120 °C silicon oil bath. The homogeneous mixture is stirred under an atmosphere of N2(g) for approximately 5 min at which point a solid precipitates from the solution. The resulting heterogeneous mixture is allowed to cool to ambient temperature with stirring and is then immersed in an ice/water bath. The resulting white precipitate is collected by suction filtration and washed with toluene (5 × 20 mL). The filtrate is concentrated using a rotary evaporator (60 °C, 15 mmHg) followed by a high vacuum (ambient temperature, <0.75 mmHg, 1 h) affording a brown solid. Toluene
is added (30 mL) and the homogeneous mixture is placed into a 60 °C water bath until heterogeneity (Note 28) is reached (approximately 3-5 min). The suspension is allowed to cool to ambient temperature with stirring. The resulting white precipitate is collected by suction filtration and washed with toluene (2 × 20 mL). The filtrate is concentrated using a rotary evaporator (60 °C, 15 mmHg) followed by high vacuum (ambient temperature, <0.75 mmHg, 1 h) affording a brown solid. Toluene
is added (10 mL) and the homogeneous mixture is placed into a 60 °C water bath until heterogeneity (Note 28) is reached again. The suspension is allowed to cool to ambient temperature with stirring. The resulting white precipitate is collected by suction filtration and washed with toluene (2 × 20 mL). The collected white solids are transferred into a 20 mL vial and residual volatiles are removed under high vacuum (ambient temperature, <1 mbar, >24 h) affording 5.54-5.89 g (60-64%) of (5aS,10bR)-5a,10b-dihydro-2-(2,4,6-trimethylphenyl)-4H,6H-indeno[2,1-b]-1,2,4-triazolo[4,3-d]-1,4-oxazinium chloride (4) as a white powder (Note 29).
2. Notes
1.
The checkers found that it was difficult to maintain stirring without the use of an overhead stirrer.
2.
The authors used an Eyela Low-Temp Pairstirrer PSL-1400 and a MeOH bath operating at -10 °C, while the checkers used a Neslab CC-100 to keep an acetone bath operating at -10 °C.
3.
Hydrogen chloride solution (ACS certified, 37%) was purchased from Fisher Scientific.
4.
Deionized water was used.
5.
2,4,6-Trimethylaniline (97%, Alfa Aesar) was fractionally distilled under reduced pressure (80 °C, 0.75 mmHg) from zinc metal (granular) prior to use.
6.
CAUTION: Due to the slight exotherm from the addition, HCl(g) evolves from the flask.7.
An additional 10 mL of deionized water was used to wash the wall of flask.
8.
Sodium nitrite (Fisher) was used as received. The addition of the aq NaNO
2 solution was carried out over a period of 45 min with an internal temperature range of -5-3 °C and resulted in a homogeneous yellow solution.
9.
Tin(II) chloride dihyrdrate (Fisher) was used as received. The dissolution of stannous chloride in 1:1 conc. HCl/H
2O is incomplete resulting in an iridescent, slightly heterogeneous solution.
10.
A 150-mL medium-porosity Büchner funnel lined with filter paper (Whatman Grade No. 3) was used. Alternatively, a similar filter funnel of fine-porosity can be used.
11.
Diethyl ether (99.9%, EMD) was used as received.
12.
The checkers found that the initial precipitate is very fine and a long time is required for the filtration. Dissolving in MeOH and re-concentration is helpful to convert the fine powder into a more crystalline material that can be easily filtered.
13.
VWR B 2500A-MTH ultrasonic cleaner with 85 W was used and ice is added occasionally to maintain the temperature below 30 °C
14.
The product had the following physiochemical properties: Mp 195-197 °C (dec.); IR (neat) ν 3296, 3002, 2964, 2911, 2691, 1564, 1515, 1479, 849, 830, 757 cm
-1;
1H NMR
pdf (400 MHz, DMSO-d
6) δ: 2.20 (s, 3 H), 2.35 (s, 6 H), 6.60 (bs, 1 H), 6.86 (s, 2 H), 9.76 (variable bs, 3 H);
13C NMR
pdf (100 MHz, DMSO-d
6) δ: 17.8, 20.4, 129.0, 134.9, 136.1, 137.9; HRMS (ESI)
m/z calcd. for C
9H
15N
2 (M
+) 151.1223; found 151.1228; Anal. calcd. for C
9H
15ClN
2: C, 57.90; H, 8.10; N, 15.01; found: C, 57.82; H, 8.19; N, 15.04.
15.
A 5/8 inch × 1 1/4 inch egg-shaped stir bar was used.
16.
(4a
R,9a
S)-4,4a,9,9a-tetrahydroindeno[2,1-
b][1,4]oxazin-3(2
H)-one was prepared from (
1R,
2S)-(-)-
cis-1-amino-2-indanol, following the preceding
Organic Syntheses procedure reported by Rovis and co-workers (
Org. Synth.
2010,
87, 350-361).
17.
Methylene chloride (99.9%) was purchased from Fischer Scientific and purified by pressure filtration under argon through activated alumina.
18.
Trimethyloxonium tetrafluoroborate (98%, Aldrich) was used as received. Direct contact of skin with trimethyloxonium tetrafluoroborate must be avoided because of its caustic nature and alkylating properties.
19.
The purity of the crude product was estimated to be about 95% based on the crude
1H NMR. An analytically pure sample can be obtained via flash column chromatography (Sorbent Silica Gel 60 (230-400 Mesh), pentane/ethyl ether 7:3) and has the following physiochemical properties: R
f = 0.42 (EMD precoated plates (silica gel 60 F254, Art 5715, 0.25 mm, pentane/ethyl ether 7:3); Mp 73-75 °C (uncorrected); [α]
D20 −15.7 (
c 1.26, EtOH); IR (neat) ν 1683, 1443, 1383, 1238, 1100 738 cm
-1;
1H NMR
pdf (600 MHz, CDCl
3) δ: 3.02 (d, 1 H,
J = 16.8 Hz), 3.20 (dd, 1 H,
J = 16.2, 4.8 Hz), 3.79 (s, 3 H), 3.95 (d, 1 H,
J = 15.8 Hz), 4.01 (dd, 1 H,
J = 15.7, 1.9 Hz), 4.29 (t, 1 H,
J = 4.8 Hz), 4.89 (d, 1 H,
J = 3.8 Hz), 7.21-7.26 (m, 3 H), 7.49 (d, 1 H,
J = 7.8 Hz);
13C NMR
pdf (150 MHz, CDCl
3) δ: 37.6, 52.2, 61.4, 62.8, 75.1, 124.6, 124.9, 126.8, 127.4, 139.1, 143.2, 161.7; HRMS (APCI)
m/z calcd. for C
12H
14NO
2 ([M+H]
+) 204.1025, found 204.1019; Anal. calcd. for C
12H
13NO
2: C, 70.92; H, 6.45; N, 6.89; found: C, 70.84; H, 6.48; N, 6.82.
20.
Freshly-opened MeOH (anhydrous, EMD Chemicals) was used.
21.
Freshly-opened EtOAc (anhydrous, EMD Chemicals) was used. The submitters reported that EtOAc was dried by passage through an alumina drying column under an atmosphere of Argon. The submitters have observed that if anhydrous EtOAc is not used a lower yield is obtained due to hydrolysis of the amidrazone intermediate.
22.
The product was contaminated with 2-mesitylhydrazinium chloride (
1), which could not be removed. The checkers and submitters typically observed ~5-10% contamination based on
1H NMR integration of the purified amidrazone hydrochloride
3.
23.
The product had the following physiochemical properties: Mp 200-201°C (dec.); [α]
D20 66.7 (
c 1.42, EtOH); IR (neat) ν 3294, 3119, 2997, 2966, 2951, 2916, 2731, 2692, 1674, 1514, 1482, 1330, 845, 739 cm
-1;
1H NMR
pdf (400 MHz, DMSO-d
6) δ: 2.20 (s, 3 H), 2.22 (s, 6 H), 2.97 (d, 1 H,
J = 16.9 Hz), 3.29 (dd, 1 H,
J = 16.9 Hz, 4.7 Hz), 4.55 (d, 1 H,
J = 16.7 Hz), 4.62 (d, 1 H,
J = 16.7 Hz), 4.72 (t, 1 H,
J = 4.4 Hz), 5.00 (t, 1 H,
J = 3.6 Hz), 6.86 (s, 2 H), 7.13 (s, 1 H), 7.27-7.32 (m, 3 H), 7.69-7.73 (m, 1 H), 11.08 (d, 1 H,
J= 3.2 Hz), 11.46 (s, 1 H);
13C NMR
pdf (100 MHz, DMSO-d
6) δ: 17.9, 20.2, 37.0, 56.1, 60.1, 76.8, 124.75, 124.83, 126.6, 128.1, 129.3, 131.2, 133.7, 138.2, 139.7, 140.2, 159.2; HRMS (ESI)
m/z calcd for C
20H
24N
3O (M
+) 322.1914; found 322.1913.
24.
Chlorobenzene (99%, Aldrich) was dried over molecular sieves (4Å) prior to use.
25.
Triethylorthoformate (98%, Acros) was used without further purification.
26.
Anhydrous hydrogen chloride (4 M dioxane) was purchased from Aldrich Chemical Co. and used as received.
27.
CH
2Cl
2 (3 × 20 mL) was used to rinse the sealed tube and transferred into the 250-mL, round-bottomed flask.
28.
As soon as toluene is added, the solid dissolves and a brown homogeneous solution is formed. Within 3-5 min of heating at 60 °C, a precipitate is formed rapidly.
29.
The product had the following physiochemical properties: Mp 217-219 °C; [α]
D20 −133.5 (
c 1.00, EtOH); IR (neat) ν 3435, 3482, 2904, 2853, 1580, 1466, 1222, 1097, 1083, 847, 749, 729, 662 cm
-1;
1H NMR
pdf (400 MHz, DMSO-d
6) δ: 2.12 (s, 6 H), 2.37 (s, 3 H), 3.16 (d, 1 H,
J = 17.0 Hz), 3.50 (dd, 1 H,
J = 16.9, 4.8 Hz), 4.99 (t, 1 H,
J = 4.4 Hz), 5.08 (d, 1 H,
J = 16.0 Hz), 5.26 (d, 1 H,
J = 16.0 Hz), 6.12 (d, 1 H,
J = 4.0 Hz), 7.21 (s, 2 H), 7.33-7.45 (m, 3 H), 7.65 (d, 1 H,
J = 7.2 Hz), 11.34 (s, 1 H);
13C NMR
pdf (100 MHz, DMSO-d
6) δ: 16.9, 20.6, 37.0, 59.7, 61.1, 76.8, 124.0, 125.3, 127.1, 129.2, 129.3, 131.2, 134.8, 136.1, 140.6, 141.3, 144.7, 150.0; HRMS (ESI)
m/z calcd. for C
21H
22N
3O (M
+) 332.1757; found 332.1756; Anal. calcd. for C
21H
24ClN
3O
2 (M+H
2O): C, 65.36; H, 6.27; N, 10.89; found: C, 65.43; H, 6.24; N, 10.88.
Safety and Waste Disposal Information All hazardous materials should be handled and disposed of in accordance with "Prudent Practices in the Laboratory"; National Academy Press; Washington, DC, 1995.
3. Discussion
In 1997, Leeper reported the design and synthesis of a new class of chiral triazolium salts derived from chiral 1,2-amino alcohols.
2 While the initial application of these precatalysts to enantioselective benzoin reactions provided only moderate selectivities, subsequent work by Rovis and Enders led to the identification of broadly useful aminoalcohol-derived catalysts that have proven useful for highly enantioselective intermolecular homobenzoin reactions,
3 intramolecular Stetter reactions,
4 and intramolecular benzoin cyclizations.
5 Of particular utility, in terms of reaction yield and enantioselectivities, is Rovis' aminoindanol-derivated triazolium precatalysts bearing
N-phenyl,
N-
para-methoxyphenyl, or
N-pentafluorophenyl substituents.
4cScheme 1. Reactive pathways promoted by N-mesityl carbene 5 (Mes = 2,4,6-trimethylphenyl).
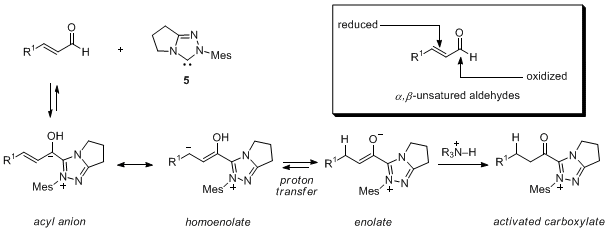
In our own studies, we have developed redox reactions of alpha-functionalized aldehydes for the catalytic generation of activated carboxylates, homoenolates, and enolates (Scheme 1). As part of these efforts, we have documented a profound, product-determining role for both the catalyst type (imadazolium vs. triazolium)
6 and the nature of the
N-substituent.
7 A series of investigations have identified a critical need for
N-mesityl substituted triazolium precatalysts for effective conversions, a finding that has been mirrored in subsequent studies by other groups on new reactions catalyzed by chiral
N-heterocyclic carbenes.
8 For example, in our own work we have found the
N-mesityl-substituted aminoindanol-derived precatalyst
4 to be particularly effective in controlling the enantioselectivity of a wide variety of novel annulation reactions.
9 A selection of processes employing
N-mesityl substituted triazolium salts reported by our group is shown in Figure 1.
Figure 1. Selected stereoselective annulations promoted by N-mesityl substituted aminoindanol-derived NHC precatalyst 4.
Rovis has described a concise, high-yielding synthesis of a number of chiral aminoindanol-derived triazolium salts.
10 While we have found these protocols to be very effective for the synthesis of triazolium salts bearing simple aromatic groups, we obtained capricious outcomes when attempting to apply these procedures to the preparation of the
N-mesityl substituted variant. In our efforts to adapt Rovis' procedures to a preparative scale synthesis of
N-mesityl substituted triazolium salts, we traced much of our difficulties to the low purity and poor stability of 2-mesitylhydrazine.
11 To circumvent this, we adopted the protocol originally reported by Knight and Leeper,
2 in which the neutral iminoether was allowed to react with the arylhydrazine hydrochloride.
This approach required a reliable, scale synthesis of 2-mesitylhydrazinium hydrochloride (
1). To achieve this we refined a Sandmeyer approach from the corresponding aniline.
12 Critical to the success of this reaction on scale is vigilant monitoring of the internal reaction temperature during diazonium formation and reduction steps by maintaining a cold (-10 °C) external bath and by portion-wise additions of sodium nitrite and stannous chloride. Although the isolated yield of this particular hydrazinium is moderate, this procedure is robust and reproducible on a preparative scale.
The condensation of 2-mesitylhydrazinium hydrochloride and the iminoether was carried out under the conditions reported by Leeper with the exception that a catalytic amount of anhydrous HCl was found to be beneficial for the reaction outcome.
13 The most challenging aspect of the synthesis of the
N-mesityl substituted triazolium salts was the ring-closing reaction with triethylorthoformate. After considerable experimentation, we found two important factors that led to clean, high-yielding reactions for the production of
N-mesityl substituted triazolium salts. First, it was essential to add an equivalent of anhydrous HCl to the reaction mixture. Second, and most critically, we found that extended reaction times were detrimental to the isolation of the desired triazolium salts. In contrast to prior protocols that employed longer reactions times (>12 hours), reaction periods of 1-2 hours were generally preferred. Qualitatively, the progress of the reaction could be followed by the solubilization of the starting amidrazone hydrochloride; when a clear solution had formed the reaction was generally complete.
This procedure is not limited to the synthesis of the aminoindanol-derived triazolium salt
4. We have also found it to be directly applicable to other classes of
N-mesityl substituted triazolium NHC precatalysts (Figure 2). We reported the first
N-mesityl-substituted triazolium salt (
5) in 2005 for the redox esterification of enals,
7 which we have conveniently named RMesCl. Further, we have demonstrated that this procedure was amenable to
N-mesityl substituted triazolium salts based on bicyclic scaffolds of increasing ring size (
614-
7), the chiral bicyclic scaffold popularized by Rovis (
8-
10),
10,15 and the achiral (
11) and chiral (
12-
13)
16 morpholinone-derived bicyclic scaffold. We anticipate that the synthesis of novel
N-mesityl substituted triazolium NHCs by our method will facilitate the discovery of new NHC-promoted processes and offer the possibility to render such processes enantioselective.
Figure 2. Examples of N-mesityl substituted triazolium salts prepared by our reported method (Mes = 2,4,6-trimethylphenyl).
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2,4,6-Trimethylaniline; (88-05-1)
Hydrogen chloride, concentrated aqueous solution 37%; (7647-01-1)
Sodium nitrite; (7632-00-0)
Tin(II) chloride dihydrate; (10025-69-1)
Trimethyloxonium tetrafluoroborate; (420-37-1)
Hydrogen chloride, dioxane solution (4 M); (7647-01-1)
2-Mesitylhydrazinium chloride; (76195-82-9)
Chlorobenzene; (108-90-7)
Triethyl orthoformate; (122-51-0)
(5aR,10bS)-5a,10b-Dihydro-2-(2,4,6-trimethylphenyl)-4H,6H-indeno[2,1-b]-1,2,4-triazolo[4,3-d]-1,4-oxazinium chloride; (903571-0 2-8)
 |
Jeffrey W. Bode was born in California in 1974 and studied chemistry and philosophy at Trinity University in San Antonio, Texas. He received his Dok. Nat. Sci. from the Eidgenössicsche Technische Hochschule (ETH) in Zürich, Switzerland with Prof. Erick M. Carreira in 2001. Following a JSPS Postdoctoral Fellowship with Prof. Keisuke Suzuki at the Tokyo Institution of Technology, he joined the faculty of the University of California, Santa Barbara as an Assistant Professor in 2003. In 2007, he joined the University of Pennsylvania in Philadelphia, Pennsylvania as an Associate Professor of Chemistry. His research interests include the development of new synthetic methods, catalysis, peptide synthesis, and bioorganic chemistry. |
 |
Dr. Justin R. Struble was born in Toledo, OH in 1981. An Ohio native, he received his B.A. in Chemistry in 2003 from Kenyon College. In 2004, he joined Dr. Jeffrey Bode at University of California Santa Barbara where he developed N-Heterocyclic Carbene (NHC) catalysts for their application in stereoselective transformations. In 2007, along with Dr. Bode, he moved to the University of Pennsylvania where he received his Ph.D. in 2009. Currently he is a postdoctoral research associate for Dr. Martin Burke at the University of Illinois Urbana-Champaign where he focuses on expanding the capabilities of MIDA boronates in metal-catalyzed cross-coupling reactions as well as working on the total synthesis of Amphotericin B and its derivatives. |
 |
Yajing Lian graduated with a B.S. degree from Xiamen University, P.R. China in August 2003 with chemistry major. After graduation, he joined Dr. Robert J. Hinkle's group at the College of William and Mary in Virginia for his Masters study, investigating the bismuth catalyzed tandem cyclization reactions and the iodonium(III) chemistry. He received his masters degree in 2005. In 2006, Yajing Joined Dr. Huw Davies' group at S.U.N.Y. at Buffalo for his Ph. D study and then he moved to Emory University in 2008 together with the whole group. Currently he is developing new reactions of rhodium carbenoid and applying these to the total synthesis of natural products. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved