Org. Synth. 2012, 89, 549-561
DOI: 10.15227/orgsyn.089.0549
Direct Synthesis of Azaheterocycles from N-Aryl/Vinyl Amides.
Synthesis of 4-(Methylthio)-2-phenylquinazoline and 4-(4-Methoxyphenyl)-2-phenylquinoline
Submitted by Omar K.
Ahmad, Jonathan William Medley, Alexis Coste, and Mohammad Movassaghi
1,2.
Checked by Hiromi Kaise and Tohru Fukuyama.
1. Procedure
4-(Methylthio)-2-phenylquinazoline (3)
.
A flame-dried, 300-mL three-necked round-bottomed flask equipped with a 3.0 cm football-shaped stir bar, rubber septum, and low temperature thermometer is charged with benzanilide (1) (Note 1) (6.02 g, 30.5 mmol, 1 equiv), sealed under an argon atmosphere, and fitted with an argon inlet.
Anhydrous dichloromethane (Note 2) (60 mL) followed by 2-chloropyridine (Note 3) (5.76 mL, 6.97 g, 61.4 mmol, 2.01 equiv) is added via syringe, and the heterogeneous mixture is vigorously stirred and cooled to <-70 °C (dry-ice-acetone bath, internal temperature).
After 10 min, trifluoromethanesulfonic anhydride (Note 4) (Tf2O, 5.60 mL, 9.39 g, 33.3 mmol, 1.09 equiv) is added via syringe over 5 min at <-65 °C (internal temperature).
After 15 min, the reaction flask is warmed to 0 °C (ice-water bath).
After 5 min, the deep red solution becomes homogeneous and a solution of thiocyanic acid methyl ester (2) (Note 1) (2.52 mL, 2.67 g, 36.5 mmol, 1.20 equiv) in anhydrous dichloromethane (40 mL) is added via cannula over 5 min at 5-6 °C (Note 5).
After 10 min, the cold bath is removed, and the reaction mixture is allowed to warm to 23 °C.
After 2.5 h, triethylamine (Note 2) (10.0 mL, 7.26 g, 71.7 mmol, 2.35 equiv) is added via syringe over one min.
The resulting mixture is concentrated with a rotary evaporator (20 mmHg, 30 °C).
The remaining deep red oil is purified by flash column chromatography (Note 6) to afford quinazoline 3 (6.15 g, 80%) as an off-white solid (Note 7).
4-(4-Methoxyphenyl)-2-phenylquinoline (5)
.
A flame-dried,
300-mL, three-necked round-bottomed flask equipped with a 3.0 cm football-shaped stir bar, rubber septum, and thermometer is charged with benzanilide (1) (Note 1) (5.29 g, 26.8 mmol, 1 equiv), sealed under an argon atmosphere, and fitted with an argon inlet.
Anhydrous dichloromethane (Note 2) (45 mL) followed by 2-chloropyridine (Note 3) (5.04 mL, 6.10 g, 53.7 mmol, 2.00 equiv) is added via syringe, and the heterogeneous mixture is vigorously stirred and cooled to <-70 °C (dry ice-acetone bath, internal temperature).
After 10 min, trifluoromethanesulfonic anhydride (Note 4) (Tf2O, 4.96 mL, 8.33 g, 29.5 mmol, 1.10 equiv) is added via syringe over 5 min at <-65 °C (internal temperature).
After 10 min, the reaction flask is warmed to 0 °C (ice-water bath).
After 10 min, the yellow solution becomes homogeneous, and a solution of p-methoxyphenylacetylene (4) (Note 1) (4.18 mL, 4.26 g, 32.2 mmol, 1.20 equiv) in anhydrous dichloromethane (45 mL) is added via cannula over 5 min at <10 °C (internal temperature).
After 2 h, aqueous saturated sodium bicarbonate solution (60 mL) is added carefully to the dark green reaction mixture, the cold bath is removed, and the reaction mixture is allowed to warm to 23 °C.
After 10 min, the reaction mixture is transferred to a 1-L separatory funnel and diluted with aqueous saturated sodium chloride solution (200 mL) and dichloromethane (200 mL), and the layers are separated.
The aqueous layer is extracted with dichloromethane (2 × 250 mL).
The combined organic layers are dried over anhydrous sodium sulfate (160 g), filtered and concentrated with a rotary evaporator under reduced pressure (20 mmHg, 30 °C).
To remove the 2-chloropyridine, the contents of the flask are quantitatively transferred with dichloromethane (~45 mL) into a 100-mL pear-shaped flask and concentrated with a rotary evaporator under reduced pressure (20 mmHg, 30 °C).
A 3.0 cm football-shaped stir bar is added to the flask, and the residue is further concentrated with agitation under reduced pressure (<1 mmHg, 23 °C) for 12 h to afford a brown oil.
The flask is placed in an oil bath while kept under reduced pressure (<1 mmHg), and the oil bath is warmed to 95 °C over 45 min.
After 3 h, the flask is removed from the oil bath and allowed to cool to 23 °C.
The residue is purified by flash column chromatography (Notes (Note 8) and (Note 9)) to afford quinoline 5 (7.34 g, 88%) as a light brown solid (Note 10).
2. Notes
1.
Benzanilide (98%) was used as purchased from Aldrich Chemical Company, Inc.
Thiocyanic acid methyl ester (99+%) was used as purchased from TCI.
p-Methoxyphenylacetylene (99%) was used as purchased from Alfa Aesar.
2.
The submitters purchased dichloromethane from J.
T.
Baker (Cycletainer
TM) and triethylamine from Aldrich Chemical Company, Inc.
Both were purified by the method of Grubbs et al.
under positive argon pressure.
3 The checkers purchased dichloromethane from Wako Pure Chemical Industries, Ltd.
(anhydrous, 99.5%) and triethylamine (>99%) was purchased from Kanto Chemical Co., Inc.
and dried over KOH.
3.
2-Chloropyridine (99%) was purchased from Aldrich Chemical Company, Inc.
and was purified by distillation from calcium hydride (26 mmHg, 72 °C) and stored sealed under an argon atmosphere.
4.
Trifluoromethanesufonic anhydride (98%) was purchased from Oakwood Products, Inc.
and used as received.
Reagent quality is important for the success of these reactions.
The checkers discovered that the use of trifluoromethanesulfonic anhydride purchased from TCI resulted in incomplete reactions and a significantly lower yield of
3 and
5 when the reagent was used without purification.
The checkers did observe that the desired products could be obtained in good yield if freshly distilled reagent from TCI was employed for the reactions.
5.
The reaction mixture became heterogeneous as precipitates formed within several minutes of thiocyanic acid methyl ester addition.
6.
Flash column chromatography (8.0 cm diameter, 10 cm height) was performed using silica gel (Silica Gel 60, spherical, 40-100 mm, (Kanto, 250 g) (submitters: 60-Å pore size, 40-63 mm, standard grade, Zeochem)).
The residue was loaded using 25% dichloromethane in
n-hexane (20 mL).
The eluent was 6% ethyl acetate in
n-hexane until quinazoline
3 (TLC:
Rf = 0.64, 20% ethyl acetate in
n-hexane (submitters: TLC:
Rf = 0.49, 20% ethyl acetate in hexanes), visualized by UV and KMnO
4 stain) completely passed through the column (fractions #9-26 collected, ~100 mL/fraction tubes).
The combined fractions were concentrated under reduced pressure (25 mmHg, 30 °C) using a rotary evaporator.
Any residual 2-chloropyridine (TLC:
Rf = 0.59, 20% ethyl acetate in
n-hexane (submitters:
Rf = 0.35, 20% ethyl acetate in hexanes), visualized by UV) can be readily removed from quinazoline
3 under reduced pressure (<1 mmHg).
TLC analysis was performed on silica gel 60 F
254 (0.25 mm, Merck) by the checkers (Submitters: EMD Silica Gel 60F plates).
7.
When trifluoromethanesufonic anhydride from TCI was distilled prior to use, a yield of 74% was observed.
The analytical data for quinazoline
3 is as follows: mp 89-90 ºC;
1H NMR
pdf(400 MHz, CDCl
3, 20 °C) δ: 2.85 (s, 3 H), 7.50-7.55 (m, 4 H), 7.83 (td, 1 H,
J = 8.2, 1.4 Hz), 8.03 (d, 1 H,
J = 9.2 Hz), 8.09 (d, 1 H,
J = 8.3 Hz), 8.64 (dd, 2 H,
J = 7.8, 2.3 Hz);
13C NMR
pdf(100 MHz, CDCl
3, 20 °C)
δ: 12.6, 122.5, 123.7, 126.6, 128.4, 128.5, 129.0, 130.5, 133.5, 138.1, 148.7, 158.8, 171.3; FTIR (neat) cm
−1: 3059, 2923, 1614, 1558, 1536, 1482, 1335, 1304; HRMS (ESI) calcd for C
15H
13N
2S [M+H]
+: 253.0799, found: 253.0805; Anal.
calcd for C
15H
11N
2S: C, 71.40; H, 4.79; N, 11.10.
Found: C, 71.47; H, 4.97; N, 11.00.
8.
Flash column chromatography (7.0 cm diameter, 16 cm height (submitters: 7.0 cm diameter, 26 cm height)) was performed using silica gel (Silica Gel 60, spherical, 40-100 mm (Kanto, ca.
320 g) (submitters: 60-Å pore size, 40-63 mm, standard grade, Zeochem)).
The residue was loaded using 25% dichloromethane in
n-hexane (20 mL).
The eluent was
n-hexane for the first five fractions (fractions #1-5, ~100 mL/fraction tubes), then 5% ethyl acetate in
n-hexane until fraction 42 (fractions #6-42, ~100 mL/fraction tubes), then 8.5% ethyl acetate in
n-hexane until fraction 89 (fractions #43-89, ~100 mL/fraction tubes), and finally 10% ethyl acetate in
n-hexane until quinoline
5 (TLC:
Rf = 0.61, 20% ethyl acetate in
n-hexane (submitters: TLC:
Rf = 0.35, 20% ethyl acetate in hexanes), visualized by UV and KMnO
4 stain) completely passed through the column.
The combined fractions (#27-110) were concentrated under reduced pressure (25 mmHg, 30 ºC) using a rotary evaporator.
Any residual 2-chloropyridine (TLC:
Rf = 0.59, 20% ethyl acetate in
n-hexane (submitters:
Rf = 0.35, 20% ethyl acetate in hexanes), visualized by UV) can be readily removed from quinoline
5 under reduced pressure (<1 mmHg).
TLC analysis was performed on EMD Silica Gel 60F plates by the submitters and on silica gel 60 F
254 (0.25 mm, Merck) by the checkers.
9.
Attempts at increasing the polarity of eluent by the checkers resulted in the contamination with 2-chloropyridine.
10.
When trifluoromethanesufonic anhydride from TCI was distilled prior to use, a yield of 73% was observed.
The analytical data for quinoline
5 is as follows: mp 96-100 °C;
1H NMR
pdf(400 MHz, CDCl
3, 20 °C) δ: 3.92 (s, 3 H), 7.09 (td, 2 H,
J = 8.7, 1.8 Hz), 7.44-7.55 (m, 6 H), 7.73 (tt, 1 H,
J = 8.2, 1.6 Hz), 7.80 (s, 1 H), 7.95 (d, 1 H,
J = 8.7 Hz), 8.19 (dd, 2 H,
J = 8.7, 1.4 Hz), 8.23 (d, 1 H,
J = 8.7 Hz);
13C NMR
pdf(100 MHz, CDCl
3, 20 °C) δ: 55.4, 114.0, 119.3, 125.6, 125.9, 126.2, 127.5, 128.8, 129.2, 129.4, 130.1, 130.7, 130.8, 139.7, 148.8, 148.9, 156.9, 159.8; FTIR (neat) cm
−1: 3060, 2825, 1608, 1592, 1516, 1502, 1492, 1249; HRMS (ESI) calcd for C
22H
18NO [M+H]
+: 312.1388, found: 312.1378; Anal.
calcd for C
22H
17NO: C, 84.86; H, 5.50; N, 4.50.
Found: C, 84.69; H, 5.73; N, 4.35.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Pyridine and pyrimidine derivatives are prevalent in natural products, pharmaceuticals, and functional materials.
4,5 The majority of conventional methods for their preparation involve the condensation of ketone or carbonyl-containing fragments with amine-containing precursors.
Based on key observations and guided by mechanistic insight in the electrophilic activation of amides, we developed methods for direct preparation of pyrimidines,
6 pyridines,
7 quinolines, quinazolines, and related derivatives.
8 The general methodology involves the direct condensative union of readily available
N-vinyl
9 or
N-aryl amides with respective nucleophiles.
Our results are consistent with the formation of a uniquely electrophilic intermediate upon amide activation with the reagent combination of trifluoromethanesulfonic anhydride and 2-chloropyridine.
The activated intermediate is subject to addition of various nucleophiles followed by cyclization to afford the corresponding azaheterocycle (Scheme 1).
6 More recent mechanistic studies have revealed the nature of the electrophilic intermediate is a function of the 2-halopyridine and amide substrate.
10 For example, while the use of 2-chloropyridine with
N-vinyl or
N-aryl amides typically results in the 2-chloropyridinium adduct, the utilization of 2-fluoropyridine with
N-alkyl or electron rich benzamides favors the corresponding nitrilium ion (Scheme 1).
Scheme 1 Plausible Mechanism for Condensative Azaheterocycle Synthesis
A variety of N-vinyl and N-aryl amides can be used with a range of nitriles as nucleophiles to form the desired pyrimidines and quinazolines (Table 1).
As described in our original report aliphatic, aromatic, and vinyl nitriles were all competent nucleophiles in this chemistry for synthesis of the corresponding pyrimidine derivatives.
Significantly, both optically active amide and nitrile substrates were used in this direct condensative pyrimidine synthesis without epimerization (Table 1, entries 2 and 3).
We recently extended this chemistry to cyanic acid-derived nucleophiles along with substrates that include carbamate, urea, or oxamate carbonyls for electrophilic activation.
8 These new substrate classes allow rapid access to densely heteroatom-substituted pyrimidine derivatives (Table 1).
Furthermore, many of these rapidly accessed products are versatile precursors for preparation of other substituted azaheterocycles that were previously inaccessible under the standard amide activation conditions due to functional group incompatibility.
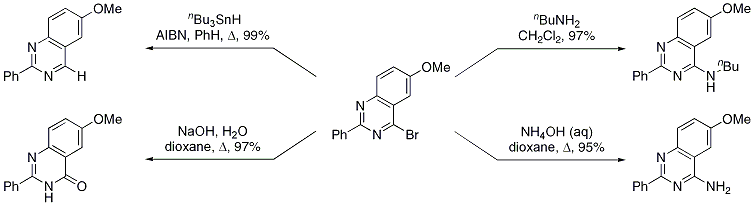
8 For example, the use of cyanogen bromide as the nitrile component (Table 1, entry 6) provides access to 4-bromopyrimidine derivatives.
Aminolysis results in 4-amino derivatives that are not accessible using the corresponding cyanamide precursors (Scheme 2).
Scheme 2 Derivatization of 4-Bromopyrimidine Derivatives
In our original communications we have reported sets of standard conditions that can be applied to a wide range of amide and nucleophile substrates.
These range from highly reactive
N-vinyl amides to less reactive electron deficient
N-aryl benzamide substrates, and from highly reactive nitriles and ynamides to less reactive enol ethers and acetylene nucleophiles (vide infra).
Most amide substrates are activated and undergo condensation with the appropriate nucleophile to the corresponding product at even sub-ambient temperatures (Scheme 3).
Typically, more forcing condensation conditions ensure further conversion to the desired product.
Pyrimidine and pyridine synthesis using
N-vinyl amides typically requires no heating and the condensation reactions are complete within minutes.
As described above, even benzanilide (
1) allows condensative azaheterocycle annulation with simple warming to room temperature.
Heating in the microwave for a short time is only necessary for the most recalcitrant substrates.
Thus the conditions required for the majority of substrates in this chemistry stand in contrast to a variety of other methods that require extended heating at elevated temperatures.
4,5Scheme 3 Condensative Azaheterocycle Synthesis at Subambient Temperatures
Furthermore, π-nucleophiles including alkoxy and silyloxy acetylenes, ynamines, ynamides, phenyl acetylenes, and vinyl ethers can all be used in conjunction with activated N-vinyl and N-aryl amide substrates to provide pyridines and quinolines, respectively (Table 2).
Interestingly, electron-rich alkynes were found to be far more reactive than vinyl ether π-nucleophiles and provided the desired heterocycles in higher yields under milder conditions.
Consistent with the observations described above in the context of pyrimidine synthesis, the condensation of an optically active amide with an ynamide provided the desired pyridine without heating (Table 2, entry 4) or loss of enantiomeric enrichment.
In the case of enol ethers an excess of the nucleophile is recommended for greater efficiency in obtaining the desired product.
In addition to the synthesis of azaheterocycles, this electrophilic activation methodology can also be used for
N-pyridinylation of amides.
10 The addition of pyridine
N-oxides to amides activated in the presence of trifluoromethanesulfonic anhydride and 2-fluoropyridine
10 as base affords the corresponding
N-pyridinyl amide in a single step (Scheme 4).
Scheme 4 Plausible Mechanism for N-Pyridylation of Amides
Table 1 Condensative Synthesis of Pyrimidine Derivatives
a Average of two experiments.
Uniform conditions unless otherwise noted: Tf2O (1.1 equiv), 2-ClPyr (1.2 equiv), nitrile (2.0 equiv), CH2Cl2, heating: A = 23 °C, 1 h; B = 45 °C, 1 h; C = microwave, 140 °C, 10 min.
D = 1,2-dichloroethane, reflux, 2 h, nitrile (4.0 equiv).
b 1.1 equiv of nitrile.
c 3.0 equiv of nitrile.
d E.e.
determined by chiral HPLC analysis of a derivative.
Table 2 Condensative Synthesis of Pyridine Derivatives
a Average of two experiments.
Uniform conditions unless otherwise noted: Tf2O (1.1 equiv), 2-ClPyr (1.2 equiv), nucleophile, CH2Cl2, heating: A = 23 °C, 1 h; B = 45 °C, 1 h; C = microwave, 140 °C, 20 min.
b nucleophile (2.0 equiv).
c 2-ClPyr (2.0 equiv).
d 10 min.
e 1 h.
f E.e.
determined by chiral HPLC analysis of a derivative.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Benzanilide: Benzamide, N-phenyl-; (93-98-1)
Dichloromethane: Methane, dichloro-; (75-09-2)
2-Chloropyridine: Pyridine, 2-chloro-; (109-09-1)
Trifluoromethanesulfonic anhydride: Methanesulfonic acid, 1,1,1-trifluoro-, 1,1'-anhydride; (358-23-6)
Thiocyanic acid methyl ester: Thiocyanic acid, methyl ester; (556-64-9)
p-Methoxyphenylacetylene: Benzene, 1-ethynyl-4-methoxy-; (26
797-70-6)
 |
Mohammad Movassaghi carried out his undergraduate research with Professor Paul A.
Bartlett at UC Berkeley, where he received his BS degree in chemistry in 1995.
Mo then joined Professor Andrew G.
Myers' group for his graduate studies and was a Roche Predoctoral Fellow at Harvard University.
In 2001, Mo joined Professor Eric N.
Jacobsen's group at Harvard University as a Damon Runyon-Walter Winchell Cancer Research Foundation postdoctoral fellow.
In 2003, he joined the chemistry faculty at MIT where his research program has focused on the total synthesis of alkaloids in concert with the discovery and development of new reactions for organic synthesis.
|
 |
Omar K.
Ahmad pursued his undergraduate studies at Brown University, where he received his Sc.B.
degree in chemistry in 2005.
As an undergraduate he worked in the laboratories of Professor Dwight Sweigart and Professor Amit Basu.
In 2005, Omar joined Professor Mohammad Movassaghi's research group at MIT for his graduate studies where his research has focused on the development of new methodologies for organic synthesis.
|
 |
Jonathan William Medley pursued his undergraduate studies at Harvard University, where he received his A.B.
degree in chemistry and physics with a minor in math in 2007.
While an undergraduate, he worked in the laboratory of Professor David A.
Evans.
In 2007, he joined the laboratory of Professor Mohammad Movassaghi at MIT for his graduate studies, which have focused on the development of new methods for organic synthesis and their application towards the total syntheses of alkaloid natural products.
|
 |
Alexis Coste carried out his graduate studies from 2007 to 2010 in the group of Professor François Couty under the supervision of Dr.
Gwilherm Evano at the University of Versailles (France) as a National Cancer Institute Fellow.
His work focused on the development of copper-catalyzed transformations and the total synthesis of the macrocyclic peptide TMC-95A.
In 2011, he joined Professor Mohammad Movassaghi's research group at MIT for pursuing postdoctoral studies where he is currently working on the total synthesis of alkaloids.
|
 |
Hiromi Kaise was born in Saitama, Japan in 1989.
She received her B.S.
in 2011 from the University of Tokyo.
In the same year, she began her graduate studies at the Graduate School of Pharmaceutical Sciences, the University of Tokyo, under the guidance of Professor Tohru Fukuyama.
Her research interests are in the area of the total synthesis of natural products. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved