1. Procedure
2. Notes
1.
Hydrazine hydrate (H
2NNH
2·1.5H
2O, 85%) was purchased from Lingfeng Shanghai by the checkers and used as received. The submitters purchased
hydrazine hydrate (H
2NNH
2·1.5H
2O, 50-60%) from Aldrich and it was used as received.
2. Use of excess
hydrazine hydrate
3.
Ethanol (=99.5%) was purchased from Zhenxing Shanghai and used as received.
4. The stirring was set to 400 rpm.
5.
2-Chlorobenzaldehyde (97%) was purchased from Alfa by the checkers and used as received.
2-Chlorobenzaldehyde (99%) was purchased by the submitters from Acros and used as received
6. Addition is mildly exothermic. Adiabatic temperature increase from 20 °C to 32 °C was observed.
7. The reaction mixture was a colorless to pale yellow solution with a small amount of yellow precipitate (bis-hydrazone
1a).
8. During the warming up period the bis-hydrazone
1a dissolved and the reaction mixture became a yellow solution. Complete decolorization was observed as the yellow colored bis-hydrazone gradually converted into the colorless mono-hydrazone
1 at 60 °C.
9. Approx. 0.1 mL sample of the reaction mixture was diluted with 0.6 mL of d
6-DMSO. Typically nearly pure mono-hydrazone
1 is observed, no bis-hydrazone
1a is detected.
10. Bath was set to 50 °C and distillation was carried out at 30-60 mmHg.
11. During the distillation product
1 separated as a liquid resulting in a milky emulsion.
12.
Methyl tert-butyl ether (99.9%) was purchased from Aldrich and used as received.
13. The extracted aqueous layer consists predominantly of
hydrazine hydrate and should be treated appropriately.
Combining with waste containing heavy metal impurities must be avoided. Dilution with copious amount of
water prior disposal is advised.
14. Combined
MTBE extracts (150 g) were colorless and opalescent.
15. Bath was set to 50 °C and distillation was carried out at 40-300 mmHg.
16. Room temperature, 2 mmHg.
17. Neat product is typically a supercooled yellowish liquid. Brief cooling of the flask results in a rapid crystallization affording a waxy solid.
18. Yield is calculated on the basis of 97% purity of the starting aldehyde. Analytical data for compound
1: mp 35.5-36.0 °C (neat); IR 3392, 3199, 1594, 1472, 1441, 1391, 1215, 1127, 1048, 1032, 914, 753, 707, 629 cm
-1;
1H NMR
pdf (400 MHz, DMSO-
d6) δ: 7.22-7.31 (m, 4 H), 7.40 (dd,
J = 7.8, 1.6 Hz, 1 H), 7.83 (dd,
J = 7.7, 1.9 Hz, 1 H), 8.06 (s, 1 H);
13C NMR
pdf (100 MHz, DMSO-
d6) δ: 125.4, 127.1, 128.4, 129.5, 130.6, 133.1, 133.5; HRMS (
m/z): [M+H
+] calcd for C
7H
8ClN
2+: 155.03705; Found 155.03738. The crude product was utilized directly in the next step without further purification.
19.
Toluene (99.8%) was purchased from Tianlian Shanghai and used as received.
20.
Potassium carbonate (98%) was purchased from Sinopharm Chemical reagent and used as received
21. The stirring was set to 400 rpm.
22.
2,4-Dichloropyridine (99%) was purchased from Energy Shanghai by the checkers and used as received.
2,4-Dichloropyridine (99%) was purchased by the submitters from Oakwood and used as received.
23. Minor bubbling was observed immediately following
2,4-dichloropyridine charge.
24.
Pd(dppf)Cl2·CH2Cl2 was purchased from Aldrich by the checkers and used as received.
Pd(dppf)Cl2·CH2Cl2 was purchased from Strem by the submitters and used as received.
25. With the addition neck capped the
nitrogen flow was set to high using bubbler as an indicator. The cap was removed and the
nitrogen flow was adjusted as necessary to maintain minimal
nitrogen flow through the bubbler.
26.
Nitrogen pressure was adjusted to maintain a visible flow through the bubbler (approx. 1 bubble per sec.).
27. Orange-pink suspension of
K2CO3 and pre-catalyst.
28. Upon reaching approximately 30 °C the suspension rapidly changed color from orange-pink to yellow. Gradual thickening of the suspension was observed as the reaction progresses. Gradual precipitation of palladium black was occasionally observed.
29. Approx. 0.1 mL sample of the supernatant was diluted with 0.6 mL of d
6-DMSO. The conversion was calculated by comparing the integration of diagnostic signals of
2,4-dichloropyridine (7.78 ppm, d, 1H) and/or
1 (8.02 ppm, s, 1H) with the integration of
toluene signal at (2.30 ppm, s, 3H). Typical conversion is =90%.
30. Efficient stirring (400-600 rpm) facilitates dissolution of inorganic by-products in the aqueous layer. Fine uniform suspension was obtained.
31. Combined filtrates consisting of dark-orange organic layer and colorless aqueous layer were discarded
32. Ambient air was passed through the wet cake overnight.
33.
DMSO (=99%) was purchased from J&K and used as received.
34. The stirring was set to 250-450 rpm.
35. The dark supernatant was allowed to fully drain using suction before
DMSO wash was introduced on the filter. The suction was cut-off and the filtercake was triturated with
DMSO using spatula and again fully drained using suction.
36. Analytical data for compound
2: mp 250-251 °C (DMSO); IR 1582, 1431, 849, 750, 704 cm
-1;
1H NMR
pdf (400 MHz, DMSO-
d6, 80 °C) δ: 6.88-6.89 (m, 1 H), 7.28 (s, 1 H), 7.35-7.41 (m, 2 H), 7.48 (d,
J = 7.3 Hz, 1 H), 8.08 (d,
J = 7.7 Hz, 1 H), 8.11 (d,
J = 5.1 Hz, 1 H), 8.43 (s, 1 H), 11.42 (s, 1 H);
13C NMR
pdf (100 MHz, DMSO-
d6, 80 °C) δ: 105.1, 114.6, 125.8, 126.8, 129.1, 129.5, 131.3, 131.5, 135.4, 143.5, 148.8, 157.3; HRMS (
m/z): [M+H
+] calcd for C
12H
10Cl
2N
3+: 266.02463; Found 266.02474.
37.
2-Methyltetrahydrofuran (98%) was purchased from Aldrich and used as received.
38. The stirring was set to 300 rpm and an off-white to grey slurry formed.
39.
Chloramine-T trihydrate (ACS reagent, 98%) was purchased from Alfa by the checkers and used as received.
Chloramine-T trihydrate (ACS reagent, 98%) was purchased by the submitters from Aldrich or Acros and used as received.
40. The internal temperature was maintained in a range from 55 to 65 °C
41. The reaction was monitored via TLC using the following method: The R
f values are 0.66 (
EtOAc/petroleum ether=1/4) for product and 0.4 (
EtOAc/petroleum ether=1/1) for starting material. The submitters monitored the reaction by HPLC by the following method: column: XBridge C18, 100x3 mm, 3.5 11m; flow rate: 0.8 mL/min; solvent A: 0.1%
trifluoroacetic acid in
water; Solvent B: 0.1%
trifluoroacetic acid in
acetonitrile; gradient: 5% B to 100% B over 12 min; wavelength: 235 nm; Retention times:
(2): 6.28 min;
(3): 6.12 min;
Chloramine-T: 6.45 min;
toluenesulfonamide: 4.34 min.
42.
Sodium sulfite (ACS reagent, ≥98.0%) was purchased from Aldrich and used as received.
43. The addition is mildly exothermic; an ice-bath may be used to control the temperature.
44. The stirring was set to 600 rpm and a homogenous biphasic mixture was obtained.
45. The additional amount of solvent was required to avoid supersaturation and crystallization of the product during aqueous work-up and charcoal-treatment.
46. A small amount of rag layer was removed with the aqueous layer.
47. The 1M
sodium hydroxide solution was prepared by dissolution of
sodium hydroxide (Sinopharm Chemical reagent, 40 g) in
water (1 L).
48. The 5M
sodium chloride solution was prepared by dissolution of
sodium chloride (Sinopharm Chemical reagent, 292 g) in
water (1 L)
49. Darco G 60 (10 mesh) was purchased from Aldrich and used as received.
50. Stirring was set a 400 rpm.
51.
2-Methyltetrahydrofuran (120 mL) was added to the flask and the solvent line was marked with a pen. This line was used as reference mark during the distillation. Subsequently the flask is emptied.
52. Celite 521 was purchased from Sinopharm Chemical reagent and used as received.
53. Depending on solvent loss during vacuum filtration and efficiency of distillate condensation solvent amounts between 460 and 580 mL were collected.
54.
Heptane (Chromasolv, =99.0%) was purchased from Tianlian Shanghai and used as received.
55. mp 140-142 °C (MeTHF/
heptane); IR 1630, 1522, 1435, 1371, 1257, 1051, 982, 936, 864, 747, 714 cm
-1;
1H NMR
pdf (400 MHz, CDCl
3) δ: 6.85 (d,
J = 7.2 Hz, 1 H), 7.49-7.51 (m, 1 H), 7.56-7.61 (m, 2 H), 7.68 (d,
J = 7.6, 1 H), 7.75 (d,
J = 7.4, 1 H), 7.85 (s, 1 H).
13C NMR
pdf (101 MHz, CDCl
3) δ: 115.1, 115.9, 124.0, 125.4, 127.6, 130.3, 132.3, 133.3, 134.0, 134.3, 145.2, 150.2; HRMS (
m/z): [M+H
+] calcd for (C
12H
8Cl
2N
3): 264.0090; Found 264.0100. Anal. Calcd. for C
12H
7Cl
2N
3: C, 54.57; H, 2.67; N, 15.91. Found: C, 54.57; H, 2.71; N, 15.87.
3. Discussion
Triazolopyridines constitute an important class of heteroaromatic compounds. The [1,2,4]triazolo[4,3-
a]pyridine moiety
2 can be found in a variety of biologically active compounds, including antibacterial, antithrombotic, antiinflammatory, antiproliferative, and herbicidal agents.3 Traditional approaches to this class of compounds rely on oxidative or dehydrative cyclizations of a linear precursor.
2 These intermediates are usually obtained through reaction of 2-hydrazinopyridines with aldehydes or acid chlorides. Depending on the underlying heterocyclic core the access to the required 2-hydrazinopyridines can be challenging.
To overcome this issue we recently described a palladium-catalyzed coupling reaction of 2-chloropyridines with aldehyde derived hydrazones (Scheme 1).
4Aldehyde-derived mono-hydrazones can be obtained in a straightforward fashion by the reaction with an excess of hydrazine.
5 The initial mixture of kinetically favored bis-hydrazone and mono-hydrazone can be readily equilibrated in the presence of an excess of hydrazine to afford the desired mono-hydrazone in nearly quantitative yields. The reaction system for the coupling is very simple and the catalyst Pd(dppf)Cl
2 is stable and readily available. Procedurally the reaction is simple and the products can be isolated by direct filtration at the end of the reaction. The oxidative cyclization step makes use of
Chloramine-T as a clean oxidant.
6 The isolation involves a simple basic aqueous work-up to remove the
toluene-sulfonamide byproduct and a charcoal treatment to remove colored trace-impurities. Crystallization is achieved from
2-methyltetrahydrofuran and
heptane.
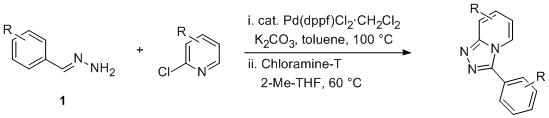
Scheme 1. Synthesis of triazolopyridines.
The reaction has a broad scope with regards to the pyridine and aldehyde component (Table 1). Additional examples have been disclosed in the original communication.
4 Satisfyingly the reaction is also suitable for other chloroazines, with a partial scope shown in Table 2, with additional examples in the original manuscript.<
4Table 1. Synthesis of triazolopyridines.
Table 2. Synthesis of related heterocycles.
The compound prepared in this procedure can be a useful building block for further functionalization. This was demonstrated by conducting a second metal-catalyzed coupling reaction on the more activated chloride substituent. Both palladium-catalyzed Suzuki-coupling
7 and iron-catalyzed coupling with a Grignard-reagent
8 afforded the desired products in good yields (Scheme 2).
Scheme 2. Further functionalization by metal-mediated coupling reactions
Appendix
Chemical Abstracts Nomenclature; (Registry Number)
Hydrazine hydrate (10217-52-4)
Benzaldehyde, 2-chloro- (35913-09-8)
Benzaldehyde, 2-chloro-, hydrazone (1) (52372-78-8)
Potassium carbonate (584-08-7)
Pyridine, 2,4-dichloro (26452-80-2)
Dichloro[1,1'-bis(diphenylphosphino)ferrocene]palladium (II) dichloromethane adduct (72287-26-4)
Benzaldehyde, 2-chloro-: 2-(4-chloro-2-pyridinyl)hydrazone (2) (1258542-95-8)
2-Methyltetrahydrofuran: Furan, tetrahydro-2-methyl- (96-47-9)
Chloramine-T trihydrate: Benzenesulfonamide, N-chloro-4-methyl-, sodium salt, hydrate (1:1:3) (9) (7080-50-4)
Sodium sulfite (7757-83-7)
1,2,4-Triazolo[4,3-a]pyridine, 7-chloro-3-(2-chlorophenyl)- (3) (1019918-88-7)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved