The drive toward increasing the fraction of
sp3 carbons in drug candidate molecules and the groundswell of academic interest in the development of visible light-driven synthetic methods has substantially increased the popularity of radical-based approaches to organic synthesis.
4,5 Studies using photoactive reagents in concert with LEDs have given rise to the highly popular field of "photoredox catalysis", which has unlocked modes of reactivity not observed in two-electron processes.
6 These odd-electron processes are not beholden to the inherent limitations of polar reactions, enabling bonds to be constructed in the presence of acidic or basic functional groups. In a watershed moment in 2014, Molander
7a and MacMillan
7b independently reported on the union of photoredox catalysis with transition metal-mediated catalysis (primarily Ni, though integration with Pd
8a and Au
8b had been previously reported). This resulted in an array of new methods for C
sp3 cross-coupling reactions (
Figure 1).
5,7c,7d,7e Indeed, industry has quickly adopted these platforms, allowing more chemical space to be surveyed on shorter timelines.
9 Recent efforts to develop continuous flow adaptations of these couplings have allowed the transition from the discovery chemistry arena to process chemistry applications.
10 The mechanistic pathways of these cross-couplings have been found to involve either Ni
0/Ni
II/Ni
III or Ni
0/NiI
I/Ni
III switches, depending on the structure of the radical involved.
11 Importantly, the origin of the radical in this process could be largely ignored so long as: (a) the photocatalyst's excited state is sufficiently oxidizing to promote substrate level oxidation and (b) the rate of homolytic fragmentation of the radical precursor aligns with the elementary cross-coupling steps of the Ni catalytic cycle. The combined efforts of several groups have resulted in the identification of a suite of carbon radical precursors for this purpose that are derived from diverse chemical feedstocks.
6,7
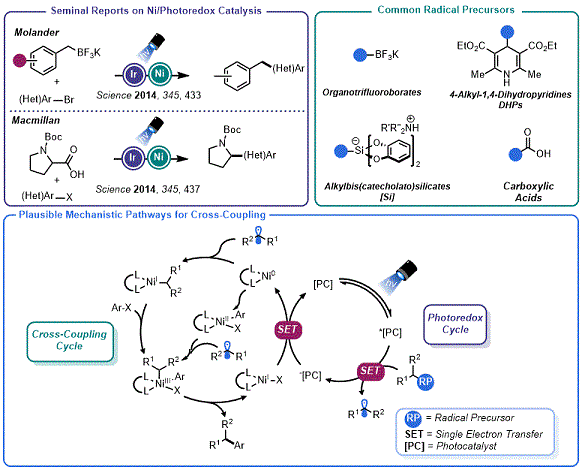
Figure 1. Overview of Ni/photoredox cross-coupling
In the mid 2010s, Molander
12a and Festerbank
12b reported a representative protocol for the synthesis of one such class of radical precursor: alkyl bis(catecholato)silicates (referred to hereafter as "organosilicates"). These reagents, which were originally investigated by Frye and co-workers in the 1960s,
13 can be easily prepared from catechol and trialkoxysilanes (typically the trimethoxy variant). These alkoxysilane precursors are widely available and remarkably inexpensive on account of their industrial-scale use in materials applications.
14 Depending on the conditions employed, organosilicates can be obtained as dialkylammonium, trialkylammonium, or potassium salts. Because of the native insolubility of the latter, 18-crown-6 is added to improve solubility in organic solvents.
12b Regardless of the counterion employed, the resulting organosilicate reagents are capable of facile photoredox-mediated oxidative homolysis to yield primary, secondary or tertiary carbon-centered radicals.
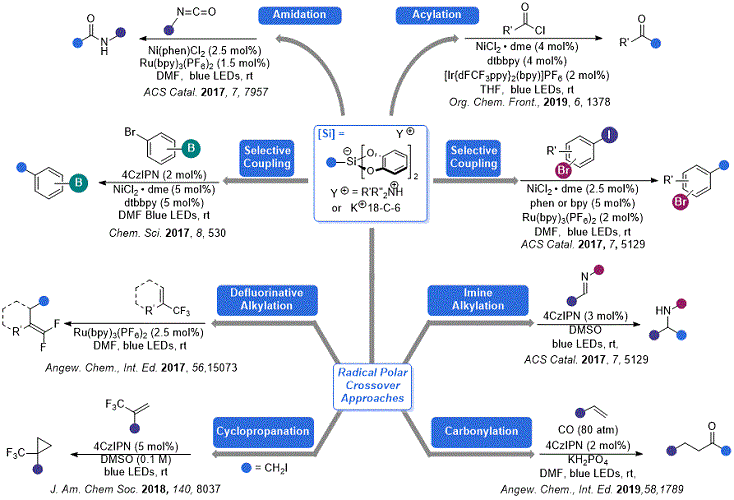
Figure 2. Some recent developments in organosilicate chemistry
As compared to their contemporary radical precursors, these organosilicate reagents have several attractive properties for photoredox applications: (1) low oxidation potentials (0.6-0.9 V) that are a function of the catechol unit rather than the alkyl fragment; (2) lack of any acidic byproducts (
i.e., BF
3) that are potentially detrimental to reaction efficiency; (3) a wide diversity of commercially available, inexpensive trialkoxysilanes that can be used to make these reagents; and (4) ease of entry into traditionally difficult-to-generate primary radicals.
15 At the time of our original
Organic Syntheses submission, only a handful of applications of these organosilicate reagents were known. Since that time, numerous examples of their use in ever more complex transformations have been reported (
Figure 2). This addendum will highlight some of the innovations in this area.
Bis(catecholato) Silicates in Radical-Polar Crossover Processes
Despite the aforementioned utility, radicals are constrained by the inherent nature of odd electron reactivity. Certain processes such as unaided addition to carbonyl species are not possible in a general sense.
16 Radical-polar crossover (RPC) processes allow synthetic chemists to tap into both homolytic and heterolytic chemistries in the same flask.
17 The crux of this approach is a single-electron transfer event that converts the radical intermediate into an ionic species without impairing the reactivity of either. Successful RPC processes allow the integration of diverse, orthogonally reactive functional groups and complex catalytic cycles. Classical variations of RPC reactions typically involved use of Kagan's reagent (SmI
2).
18 However, its high cost, strong reduction potential, and oxygen sensitivity limits its widespread synthetic utility. Photoredox catalysis enables the RPC model to be a more powerful tool for synthesis by allowing for the judicious selection of reagents with appropriate redox potentials, which enables a broad palette of transformations. Two "modes" of RPC are accessible using photoredox catalysis, depending on the polar intermediate involved (
Figure 3). In
Mode 1, the RPC process is initiated by a single electron oxidation of a substrate to give an alkyl radical, which then engages in the desired radical reaction. The resulting adduct undergoes a reduction to give an anion which can engage an electrophile in a polar step. In
Mode 2, the order of electron transfer is reversed. The first step is a single electron reduction to yield an alkyl radical which can still engage in odd-electron reactivity. Single electron oxidation then produces a cationic species that can be captured by a nucleophile. Both modes can either be net-neutral (
i.e., no external oxidant or reductant is needed) or net-reductive/oxidative (
e.g., use of Hantzch ester or DIPEA as electron sources for a reduction step).
17 In terms of organosilicate chemistry, only
Mode 1-type processes have been reported (detailed below).
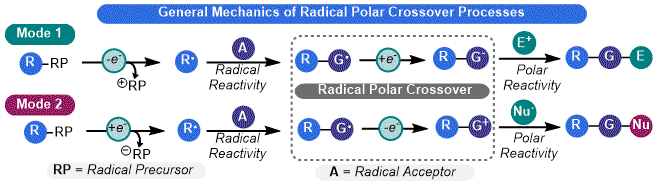
Figure 3. General modes of RPC
Radical Polar Crossover Enables Defluorinative Alkylation
In 2015 Fensterbank, Ollivier, and co-workers demonstrated that organosilicates were competent reagents in photoredox-mediated Giese addition with an array of alkenes.
12b Mechanistically, this proceeds
via a RPC event, with the polar aspect of this process being the protonation of the resulting carbanion. Although their seminal work in this area used an Ir-based photocatalyst, they later reported that the organophotocatalyst 4CzIPN
19 is also effective.
20a The authors also demonstrated that a radical addition/elimination mechanism was possible when using allylic sulfones as Giese acceptors. Derat, Ollivier, and Fensterbank also demonstrated that aryl organosilicates could also be used in this radical alkylation process.
20b With this precedent in mind, Molander and co-workers used organosilicate reagents in RPC olefin functionalization reactions. Their first success was the development of a defluorinative alkylation (DFA) protocol. Using perfluoroalkyl substituted alkenes, Molander demonstrated that radicals derived from organosilicates (and other radical precursors) readily add into these olefins yielding destabilized α-perfluoroalkyl radicals, which are prone to SET reduction (
Figure 4, top).
21 From there, facile E
1cB-type elimination yields the
gem-difluoroalkene or related species, thus constituting the polar step of the RPC process. The three radical progenitors used in this report (α-silylamines, organotrifluoroborates, and organosilicates) provided access to an array of
gem-difluoroalkenes, with organosilicates being apt for the generation of non-stabilized primary radicals. This process was extended to aryl radicals generated by halogen atom abstraction.
22
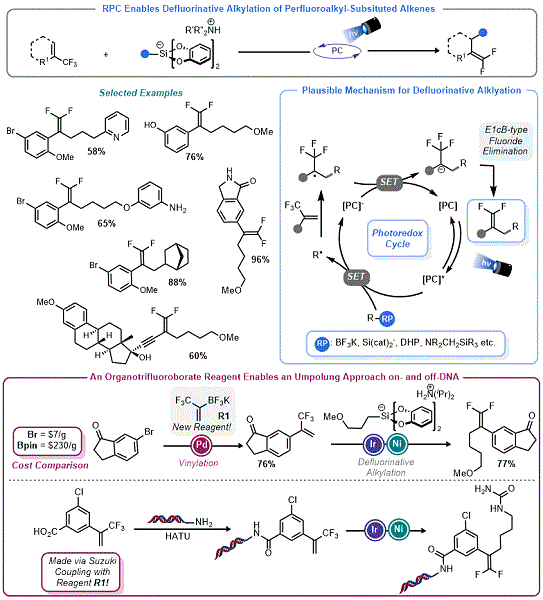
Figure 4. Defluorinative alkylation (DFA) via RPC
As part of this report
21 and a follow up paper,
23 Molander and co-workers described a new organotrifluoroborate reagent,
R1, for the rapid assembly of the trifluoromethyl-substituted styrenes used in these DFA reactions. This reagent was key for the synthesis of more complex CF
3 alkenes which would later be used as part of a larger study interfacing DNA-Encoded Library (DEL) technology with photoredox processes (
Figure 4, bottom).
24 In a collaboration with Glaxo-Smith-Kline, Molander and co-workers demonstrated that the DFA process could be executed with organosilicates in a mixture of DMSO/H
2O using several on-DNA trifluoromethyl substituted alkenes. The success of organosilicates here was surprising given that they generally degrade in aqueous organic mixtures
via hydrolysis. It is likely that the rate of radical addition and the large excess of organosilicates used in these "on DNA reactions" helped overcome this limitation.
Cyclopropanation via Organosilicate Mediated Radical Polar Crossover
Seeking to further advance the RPC model using organosilicates, Molander and co-workers sought to interrupt the E
1cB process observed in the aforementioned DFA to enable radical polar annulation reactions (RPARs). More specifically, they sought to develop a platform for the construction of cyclopropanes
via radical intermediates. The use of radicals in cyclopropanation reactions was not without precedent, but the prior art developed at the time of Molander's work was contingent on a radical based ring closure. This precedent reported by Suero and co-workers used non-redox neutral SET reduction of CH
2I
2 to generate an iodomethyl radical.
25 The latter odd-electron species could engage in Giese type addition into olefins, ultimately leading to S
H2-type ring closure to effect cyclopropanation. In contrast, Molander's approach focused on the development of a redox neutral method for cyclopropanation using a photooxidizable C
1 reagent, triethylammonium bis(catecholato)iodomethylsilicate,
R2 (
Figure 5).
26 This bifunctional reagent
27 and related species detailed later in this addendum demonstrate a unique advantage of organosilicates: the capability to easily prepare derivatives that are precursors of halogen-containing primary alkyl radicals which can be used as handles
in situ ring closure (as detailed below).
R2 was prepared in a two-step process from inexpensive chloromethyltrimethoxysilane
via a Finkelstein reaction using a protocol adapted from the parent article of this addendum. This reagent undergoes SET oxidation to produce an iodomethyl radical that engages in Giese addition onto a suitable olefin. However, since the redox environment of the resulting radical is drastically different (
i.e., the first step produced a now strongly reducing species). As such, the Giese adduct undergoes SET reduction to produce an anion which is primed for 3-
exo-tet ring closure. Although the initial focus of this work was on the generation of a library of trifluoromethylsubstituted cyclopropanes, the reaction was expanded to an array of Giese-type acceptors and styrenes. Following this report, the laboratories of Jin and Fang reported on a similar approach using potassium halomethylsilicates and activated olefins (such as vinyl silanes,
28a boronates,
28b and phosphates
28c).
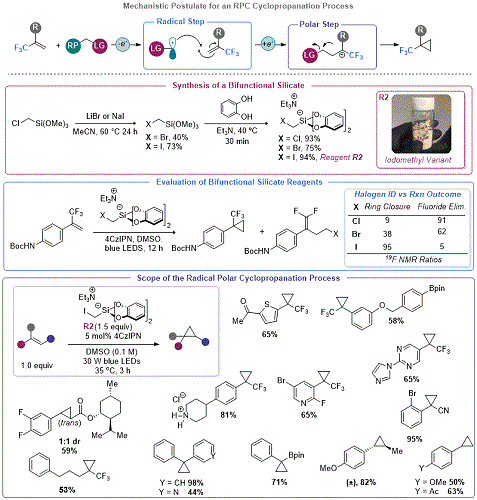
Figure 5. Development of an organosilicate cyclopropanation reagent
Bromomethyl organosilicate reagents have also been shown to be effective in cyclopropanation reactions.
28d Although
R2 was enabling for facile olefin cyclopropanation, a more general approach for organosilicate-mediated cyclopropane construction was pursued. Rather than building the electrophilic component of the RPAR process into the organosilicate itself (thus restricting the process to the installation of a methylene unit), the olefin containing species could be appended with a leaving group. Using this approach,
any photocatalytically generated radical would be able to induce cyclization upon radical addition and single-electron reduction, presuming a redox neutral photocatalytic platform was employed (
Figure 6). Using a homoallylic tosylate (assembled using a Suzuki reaction between the corresponding vinyl bromide and a borylated arene), Molander, Kelly, and co-workers identified conditions that enabled facile RPAR-type cyclopropanation using a propylacetoxysilicate.
29 The identified conditions were not a far departure from the conditions used with
R2, but a higher dilution factor seemed to improve yields. An array of organosilicates and homoallyic tosylates were competent in this reaction, affording good yields of the corresponding 1,1-disubstituted cyclopropanes. To support their conjecture that the origin of the radical was independent of the success of cyclopropanation, the authors demonstrated that other radical precursors such as organotrifluoroborates and dihydropyridines were also amenable to RPAR cyclopropanation. Likewise, Aggarwal and co-workers also disclosed a report verifying the feasibility of this approach with carboxylic acid radical precursors.
30 In a subsequent publication, Molander and Kelly demonstrated that organosilicates (and other radical progenitors) could be used in RPAR cyclopropanation with cycloalkane-embedded homoallylic tosylates to assemble polycyclic cyclopropanes.
31 This report used an array of organosilicates and proceeded in good yield with a various homoallylic tosylates, including those derived from heterocyclic systems.
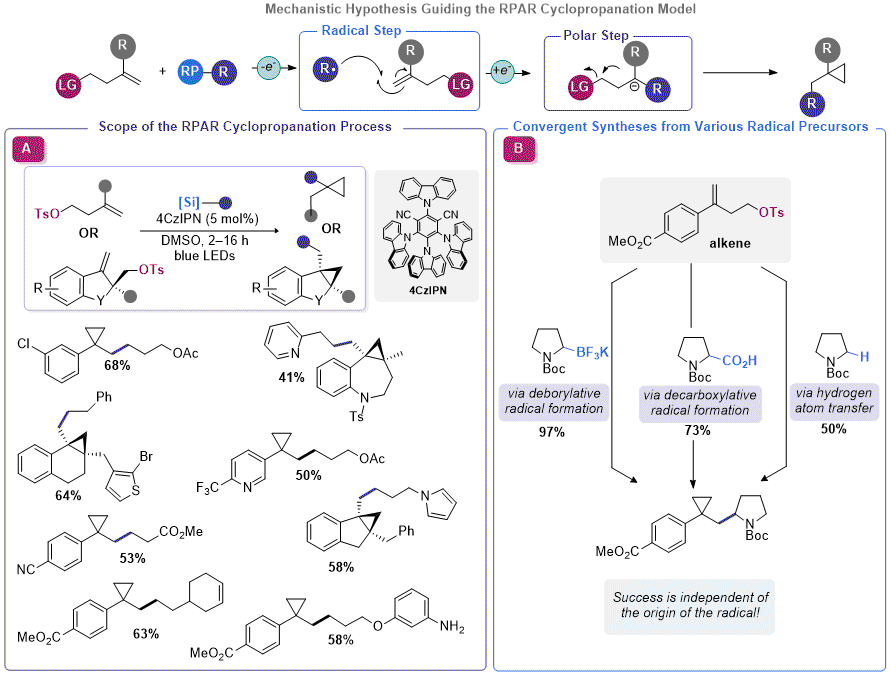
Figure 6. Utilizing the RPAR model for cyclopropanation of olefins
Engaging Imines in Organosilicate Mediated RPC
The success in employing organosilicate reagents in Giese-type additions to polarized alkenes motivated Molander and co-workers to explore addition reactions to aldimines. In 2017, they reported that irradiating an imine with a slight excess of organosilicate reagent in the presence of 4CzIPN resulted in facile alkylation.
32 This method serves as a mild, highly functional group-tolerant alternative to classical approaches for secondary amine synthesis, such as organometallic additions to aldimines or reductive amination. Molander and co-workers later expanded upon this work by using imines in an RPAR-type process.
33 A bifunctional 3-bromopropylsilicate reagent was effective in converting a range of aldimines to the corresponding pyrrolidines and piperidines (
Figure 7A).
N-Aryl imines were most effective in the transformation, whereas
N-sulfonyl imines were less effective. This may suggest that photoexcitation of the imine substrate or formation of an electron donor-acceptor (EDA) complex with the organosilicate reagent may be promoting the reaction.
34 A limitation of this work is the need for aromatic aldimines. In attempts to expand the scope of imines, Friestad demonstrated that MgCl
2 can be used as a Lewis acid additive for the radical alkylation of hydrazones derived from both aromatic and aliphatic aldehydes.
35
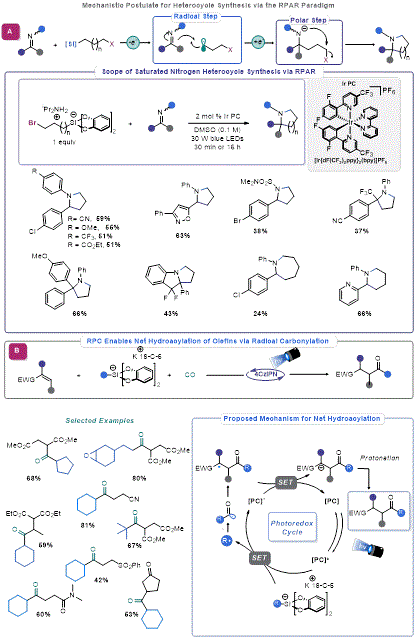
Figure 7. (a) RPC-mediated carbonylation (b) Saturated nitrogen heterocycles via RPAR
Carbonylation via Radical Polar Crossover with Organosilicates
Carbon monoxide (CO) is an established radical acceptor,
36 thus, running radical additions under high pressure of carbon monoxide enables acyl radical generation. Fensterbank exploited this propensity and the ability of acyl radicals to engage in Giese-type addition for an elegant synthesis of unsymmetrical ketones (
Figure 7B).
37 The reaction uses organosilicate-derived alkyl radicals to add to CO, after which the resultant acyl radicals engage activated olefins in Giese addition. A final SET allows for the regeneration of the photocatalyst and the desired product by protonation. A range of Giese acceptors and alkyl radicals were tolerated in this process.
Advances using Ni/Photoredox Dual Catalysis
Since the publication of the parent
Organic Syntheses protocol, advances have been made when using organosilicates in Ni/photoredox cross-coupling. For example, Vertex reported
38 a flow chemistry approach wherein reaction times could be substantially shortened in these types of cross-coupling reactions, presumably due to improved light penetration.
10Other Electrophiles: Some innovations have been disclosed with respect to types of electrophilic partners used with organosilicates. Fensterbank and Ollivier recently described a protocol for cross-coupling with 1,1-dichlorostyrenes in a stereoselective manner.
39 The conditions used in their approach were in accord with prior reports, wherein 4CzIPN in conjunction with a simple Ni/dtbbpy system were employed. A near identical system was used by Molander to selectively engage borylated aryl halides in cross-coupling.
40 Here, the authors demonstrated that C
sp2-C
sp3 cross coupling could be executed at the halogenated site and paired with a downstream Pd-mediated Suzuki-type coupling. Molander also explored the possibility of executing a "haloselective" cross-coupling using polyfunctional linchpins. In their report, a Ni/Ru metallophotoredox process for the chemoselective C
sp2-C
sp3 cross coupling of bromo(iodo)arenes using organosilicates is described.
41 Optimization revealed that phenanthroline (phen) and 2,2′-bipyridine gave the best selectivity and yield. The authors demonstrated that these polyhalogenated systems could be sequentially functionalized without need to isolate the intermediate mono-alkylated product; that is, two distinct C
sp2-C
sp3 bonds could be forged in one pot. Lastly, acyl chlorides were competent electrophiles in this paradigm, affording the desired asymmetrical ketones in moderate to good yields.
42
Although most of the literature surrounding organosilicates in Ni/photoredox cross-coupling relates to C
sp2-C
sp3 linkages, Fensterbank, Ollivier, and co-workers disclosed an approach to C
sp3-C
sp3 bond formation.
43 One of the major challenges with this type of cross-coupling is the inherent competition between homocoupling of the electrophilic partner (or radical-radical homocoupling itself). Although unable to find conditions to suppress these undesirable pathways, the authors identified conditions which afforded usable yields of primary and secondary alkyl bromides with primary and secondary alkyl radicals (derived from silicates). In this case, a Ni
0 source was necessary, as was a Ir-based photocatalyst.
Figure 8. Ni-mediated olefin dicarbofunctionalization
Dicarbofunctionalization of Olefins: Nevado and co-workers successfully explored the merger of radical addition reactions with photoredox/Ni dual cross-couplings, yielding a synthetically useful three-component dicarbofunctionalization of olefins.
44 A key factor for this transformation was the ratio between alkene and alkyl organosilicate, where an excess of either favors the more traditional difunctionalized product. As often observed with alkyl organosilicates, the functional group tolerance was excellent. This reaction was further extended to other radical precursors to give functionalized products such as unsymmetrical sulfones.
Figure 9. Amide construction via Ni/photoredox dual catalysis
Amidation via Isocyanates: Alternative ways of making amides are welcomed by the chemistry community due to the ubiquity of this bond. Molander reported an approach to the construction of amides via Ni-mediated additions onto isocyanates (
Figure 9).
45 This reaction required several modifications to more traditional alkyl organosilicate transformations, such as an alternative ligand (1,10-phenanthroline) as well as a tertiary ammonium counterion (to suppress isocyanate degradation). In contrast with other radical additions onto isocyanates, this protocol is characterized by mild conditions and the absence of a stoichiometric reductant, affording both alkyl/alkyl and alkyl/aryl amides in moderate to good yields.
Thioetherification: Although organosilicate-mediated thioetherification of aryl halides was described in the parent
Organic Syntheses protocol,
46 Molander and Petersson subsequently leveraged this reaction for modification of thiols relevant to chemical biology (
Figure 10).
47 Initially targeting arenes that could serve as biological probes or unprotected, biologically-derived thiols (such as tiopronin, glutathione, etc.), the true aim of this work was site-specific functionalization of unprotected cysteine residues on peptides. To demonstrate the feasibility of this objective, the authors performed a study wherein different amino acids were added to the standard thioetherification conditions to determine what, if any, impact they had on catalysis. In nearly all cases, these additives were tolerated up to 1 equiv. The authors showed that nonamer peptide with an internal Cys residue could be chemoselectively arylated in under 90 min in in solution phase (DMF) (at a ten-fold dilution factor).
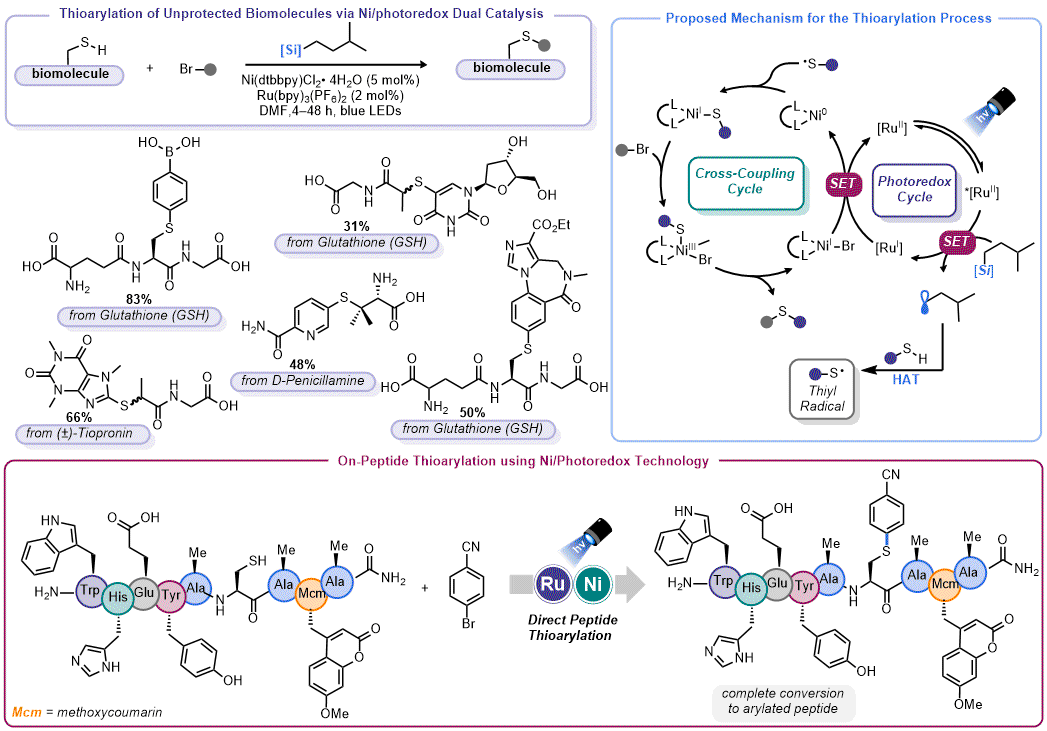
Figure 10. Organosilicate-mediated thioarylation of biomolecules
Concluding Remarks
Organosilicates like the one described in the parent article to this Addendum are powerful reagents for C-C bond construction. Using photocatalysis, these pentacoordinate silicon species can be oxidized to yield alkyl radicals via C-Si homolysis. These radicals can be funneled into RPC regimes or intercepted by low valent nickel complexes for cross-coupling protocols. Furthermore, alkyl radicals generated in this process can be used for H-atom transfer reactions with thiols to yield thiyl radicals. In addition, the simple yet robust nature of this class of radical precursor allowed for the development of a new reagent for cyclopropanation of olefins. It is anticipated that as our understanding of radical processes advance, so too will the opportunities for organosilicates to advance the state of the art in chemical synthesis.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved