Org. Synth. 2002, 79, 228
DOI: 10.15227/orgsyn.079.0228
N-HYDROXY-4-(p-CHLOROPHENYL)THIAZOLE-2(3H)-THIONE
[
2(3H)-Thiazolethione, 4-(4-chlorophenyl)-3-hydroxy-
]
Submitted by Jens Hartung and Michaela Schwarz
1
.
Checked by Raghuram S. Tangirala and Dennis P. Curran.
1. Procedure
A.
ω-Bromo-p-chloroacetophenone
oxime
. A 500-mL, four-necked, round-bottomed
flask equipped with a dropping funnel (closed with
a glass stopper), mechanical stirrer,
drying tube (
calcium chloride,
CaCl2), and a thermometer is charged
with
p-chloroacetophenone (38.6
g, 0.25 mol), glacial acetic
acid (220 mL) and aqueous
hydrobromic acid
(HBr) [1 mL, 48% (w/w),
Note 1]. The flask is immersed
in a water bath (15°C, Note 2) and
bromine
(40.0 g, 0.25 mol)
is added from the dropping funnel at such a rate that the temperature
of the reaction mixture does not exceed 25°C (Note 3). Stirring
is continued for 1 hr at 15°C. The suspension is poured onto 1 kg of crushed ice.
The colorless precipitate is collected by suction (Buchner funnel)
and repeatedly washed with small portions of water (total of 200 mL). Approximately
84 g of air-dried
ω-bromo-p-chloroacetophenone
is obtained from this step (Note 4). The crude product is transferred
into a 1-L beaker that is equipped with a magnetic
stirring bar
(Note 5). Aqueous
ethanol (96%, v/v, 400 mL)
is added and the slurry is treated with a solution of
hydroxylamine
hydrochloride
[21.7 g, 0.31 mol, (Note 6)]
in water (65 mL) in one portion at 20°C. Stirring is continued for 24 hr at 20°C
to afford a clear, pale yellow solution that is poured onto a mixture of ice (900
g) and water (400 mL).
ω-Bromo-p-chloroacetophenone
oxime
separates as a colorless solid that is collected by filtration on a Buchner
funnel. The precipitate is washed with small portions of water (total
of 200 mL) and dried for 48 hr at 20°C and 10−3 mbar (7.5 ×
10−4 mm) to furnish 53 g
(85.3%) of
ω-bromo-p-chloroacetophenone
oxime
as a colorless powder (Note 7).
B.
O-Ethyl S-[oximino-2-(p-chlorophenyl)ethyl]dithiocarbonate
. A 1-L, round-bottomed
flask equipped with a dropping funnel is charged
with a magnetic stirring bar,
potassium
O-ethyl xanthate (35.3 g, 0.22 mol),
and
acetone (140 mL).
A solution of
ω-bromo-p-chloroacetophenone
oxime (49.5 g, 0.20 mol) in
acetone (120 mL) is
added dropwise at room temperature over a period of 30 min. Stirring is continued
for 3 hr at 20°C whereupon a cloudy orange solution forms from the initial slurry.
Solids are removed by filtration using a thin pad of diatomaceous earth (1-cm height
in a Buchner funnel) to afford a clear orange solution. The
solids on the funnel are washed with
acetone
(total of 50 mL). The combined washings and filtrate are
concentrated under an aspirator vacuum to dryness to furnish
a yellow residue that is dissolved in
diethyl ether
(750 mL). This solution is washed with water (200 mL),
dried (magnesium sulfate, MgSO4),
and evaporated to dryness to afford 51.9
g (89.5%) of
O-ethyl S-[2-oximino-2-(p-chlorophenyl)ethyl]dithiocarbonate
as a yellow amorphous solid.
C.
N-Hydroxy-4-(p-chlorophenyl)thiazole-2(3H)-thione
.
O-Ethyl
S-[2-oximino-2-(p-chlorophenyl)ethyl]dithiocarbonate (56.0 g,
0.19 mol) is placed in a 500-mL round-bottomed
flask that is equipped with a magnetic stir bar.
Diethyl ether (120 mL)
is added and the slurry is treated at 0°C in small portions with solid anhydrous zinc chloride
, ZnCl2, 79.1 g, 0.58
mol) at such a rate that the solvent does not boil constantly (Note 8). After the addition is complete, the flask is stoppered with
a drying tube (CaCl2) and stirring is continued
for 48 hr at 20°C. The reaction mixture turns into a clear, dark brown solution
that solidifies toward the end of the reaction. The flask is immersed in an ice
bath and treated dropwise with 5.5 M hydrochloric
acid (140 mL, Note 9).
The precipitate dissolves immediately. Stirring is continued for 30 min at 0°C
whereupon a tan-colored solid separates. This material is collected by filtration.
It is washed with small portions of
diethyl ether
(total of 110 mL) and dried to afford 39.8 g (86%)
of
N-hydroxy-4-(p-chlorophenyl)thiazole-2(3H)-thione
(Note 10). The crude material is transferred to a 2-L,
round-bottomed flask equipped with a reflux condenser.
2-Propanol (760 mL)
is added and the reaction mixture is heated to reflux. Once a clear solution is obtained
the heat source is immediately removed (Note 11). The solution
is allowed to cool to room temperature. Precipitation of
N-hydroxy-4-(p-chlorophenyl)thiazole-2(3H)-thione
is completed by immersing the flask for 30 min in an acetone-dry ice bath
(−78°C). The product is collected by filtration and dried to afford 21.9 g (53.5%)
of
N-hydroxy-4-(p-chlorophenyl)thiazole-2(3H)-thione
as tan crystals (Notes 12, 13).
2. Notes
1.
A
three-necked, round-bottomed flask with Claisen head
may be used instead.
p-Chloroacetophenone
(97%), hydroxylamine hydrochloride (purum
p.a. ≥98%), potassium O-ethyl xanthogenate (≥98%),
bromine (puriss. p.a., ≥99.5%), and hydrobromic
acid [puriss. 48% (w/w)] were obtained from Fluka
Chemika
and used as received. All solvents [
acetic
acid (reagent grade, 99-100%, Merck &
Company, Inc.
),
diethyl ether
(purum, ≥99%, stabilized with 0.0001% of
2,6-di-tert-butyl-p-cresol, Fluka Chemika),
acetone (purum, ≥99%,
Fluka Chemika),
2-propanol
(purum, ≥99%, Fluka Chemika),
and
ethanol (purum, 96%,
2%
ethyl methyl ketone, and 0.5%
isobutyl methyl ketone, Fluka Chemika)]
were used without further purification.
Zinc chloride
was used as received from Riedel de Haen (puriss., 98-100%).
2.
The temperature of the reaction mixture is kept at 15°C throughout
the reaction by adding portions of crushed ice to the
water bath.
If the temperature drops below 10°C the solvent starts to solidify; above 25°C
products of dibromination are formed.
3.
Bromine
should be handled in a well-ventilated hood. Addition of
bromine
usually is complete within 75 min.
4.
ω-Bromo-p-chloroacetophenone
was used without further drying in the oxime-forming reaction. Drying of this product
from step (A) at 20°C and 10
−3 mbar (7.5 × 10
−4
mm) affords
58.3 g of
ω-bromo-p-chloroacetophenone
.
ω-Bromoacetophenones
are lachrymators. Skin contact should be avoided.
5.
A
4-cm stirring bar is adequate.
6.
The two reactions in Step A were taken from the literature
2
3
and were improved for the present synthesis. All previously related syntheses of arylacetophenone
oximes found in the literature have used a threefold excess of
hydroxylamine
hydrochloride
that is, however, not necessary in the present
protocol.
7.
Drying
ω-bromo-p-chloroacetophenone
oxime
was carried out in the dark since otherwise this material
turns pink in light. Wrapping the flask with aluminum foil provides adequate light
protection for this purpose.
8.
Addition of ZnCl
2 is exothermic. The reaction mixture
may bubble upon addition of each portion of ZnCl
2, but it should not boil
constantly. After addition of ZnCl
2 is complete, a
4.8-5
M solution of ZnCl2
is obtained that leads to a maximum yield
of the title compound. It was essential to use
diethyl
ether
as solvent in this step. Also no excess
diethyl
ether
should be added after the addition of ZnCl
2
is complete.
9.
5.5 M Hydrochloric acid
was prepared from
80 mL of concentrated
hydrochloric acid
and 60 mL of water. This step should
be carried out in a well-ventilated hood since an unpleasant smell develops.
10.
To some people,
N-hydroxy-4-(p-chlorophenyl)-thiazole-2(3H)-thione
has a musty odor. Thus, the submitters recommend that the compound be handled and
stored in a ventilated place. All drying operations in Step C were carried out at
20°C/10
−2 mbar (7.5 × 10
−3 mm). If prolonged
storage of the title compound is required, the use of an amber-colored vial is recommended.
11.
The solution should not be heated longer than necessary to obtain
a clear solution. According to differential thermal analysis, neat
N-hydroxy-4-(p-chlorophenyl)thiazole-2(3H)-thione
decomposes at 138 ± 2°C without melting.
12.
Analytical data for
N-hydroxy-4-(p-chlorophenyl)thiazole-2(3H)-thione
is as follows: IR (CCl
4) cm
−1:
3124, 2942, 1603, 1581, 1556,
1500, 1488, 1404, 1355, 1301,
1214, 1174, 1095, 1062, 1016,
976
; UV/Vis (EtOH)
λ
max (log ε): 309 (4.16), 240 nm (4.20)
;
1H NMR (250 MHz,
CDCl
3) δ: 6.68 (s, 1 H), 7.47 (m
c,
2 H), 7.62 (m
c, 2 H), 11.72 (br s, 1 H, OH)
;
13C NMR (100
MHz, (DMSO-d
6) δ: 106.4, 127.6, 128.7,
130.1, 134.4, 140.3, 179.2
;
Anal. Calcd for C
9H
6ClNOS
2 (243.7): C, 44.35; H,
2.48; N, 5.75; S, 26.31. Found: C, 44.35; H, 2.47; N, 5.74; S, 26.39.
13.
The submitters report that a second crop of the title compound
(
7.7 g,
16%) of similar quality separates from the mother liquor
upon concentration of the volume of the solution to 100 mL and storage at −20°C
overnight. This was not checked.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
N-Hydroxy-4-(p-chlorophenyl)thiazole-2(3H)-thione
(
1) is a valuable starting material for the synthesis of N-alkoxy derivatives
2 by a number of well-elaborated O-alkylation procedures (Figure 1).
4
5
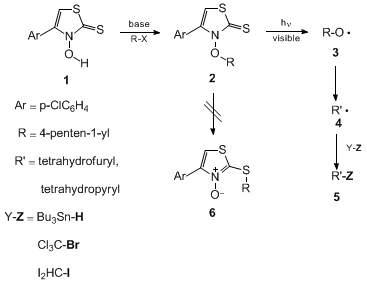
Substituted heterocycles
2 serve as sources of oxygen-centered radicals
3 for mechanistic and synthetic purposes. This method was recently applied
as a key step in the synthesis of a muscarine alkaloid.
6
N-Alkoxy-4-(p-chlorophenyl)thiazole-2(3H)-thiones
2 offer significant advantages
compared with their best current alternatives, the N-alkoxypyridine-2(1H)-thiones
7
8
since they combine two rare properties required for efficient radical precursors.
On the one hand they are sufficiently stable to allow safe handling and storage in
standard glassware without being prone to photochemical decomposition or to thermal
rearrangement,
2 →
6. This O-alkyl → S-alkyl shift is a significant
decomposition pathway for substituted N-benzylpyridine-2(1H)-thiones.
9
In contrast to their thermal stability, thiazolethiones
2 efficiently liberate
oxygen-centered radicals
3 in chain reactions upon photolysis.
4 In this sense, N-alkoxy-4-(p-chlorophenyl)thiazole-2(3H)-thiones
are superior to many existing alkoxyl radical precursors.
A smaller scale and less efficient synthesis of the phenyl derivative of
1
has been reported.
10
The method described for the title compound
1 provides excellent synthetic
access to the p-chlorophenyl-substituted thiazolethione
1. It offers higher
yields in every step of the synthesis, reduces the amount of
hydroxylamine
hydrochloride
used in Step A to a third and avoids the use of
halogenated solvents and methanol. Further, only one purification step is necessary
at the very end of the synthesis to afford pure thione
1. The protocol for
N-hydroxy-4-(p-chlorophenyl) thiazole-2(3H)-thione
has also been applied to syntheses of the respective p-substituted phenyl derivatives
7-
9
4 from the respective acetophenones
(Figure 2)
|
|
X
|
Compound
|
Yield
|
|
|
OCH3
|
7
|
46%
|
|
CH3
|
8
|
49%
|
|
Cl
|
1
|
67%
|
|
NO2
|
9
|
42%
|
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
N-Hydroxy-4-(p-chlorophenyl)thiazole-2(3H)-thione:
2(3H)-Thiazolethione, 4-(4-chlorophenyl)-3-hydroxy- (11); (105922-93-8)
ω-Bromo-p-chloroacetophenone oxime:
Ethanone,
2-bromo-1-(4-chlorophenyl)- oxime (12); (136978-96-6)
p-Chloroacetophenone:
Ethanone, 1-(4-chlorophenyl)-
(8,9); (99-91-2)
Hydrobromic acid (8,9); (10035-10-6)
Bromine (8,9); (7726-95-6)
Hydroxylamine hydrochoride (8);
Hydroxylamine,
hydrochloride (9); (5470-11-1)
O-Ethyl S-[oximino-2-(p-chlorophenyl)ethyl]dithiocarbonate:
Carbonodithioic acid, S-[2-(4-chlorophenyl)-2-(hydroximino)ethyl], O-ethyl
ester (14); (195213-53-7)
Potassium O-ethyl xanthate or
Potassium O-ethyl
xanthogenate:
Carbonodithioic acid O-ethyl ester, potassium salt
(8,9); (140-89-6)
Zinc chloride (8,9); (7646-85-7)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved