A review on the dehydrative functionalization of various phosphorus species with alcohols was published in 2019.
2 Various methods exist for the preparation of allylic
H-phosphinates, including allylation of a hypophosphorous acid (H
3PO
2) equivalent with allylic halides.
3 However, this approach is not atom-economical, requires a base, and the yields are generally moderate. In 2006, we introduced the palladium-catalyzed direct dehydrative allylation of hypophosphorous acid with allylic alcohols,
4 on which was based the 2008
Organic Syntheses article.
5 Also in 2008 was published a full paper, which dealt in part with this reaction.
6 A great feature of the original reaction is since the product is an acid, a simple extractive work-up can be employed to give products of generally high purity. However, in many instances the
H-phosphinate ester product may be more desirable for subsequent reactions. Whereas the Dean-Stark esterification of
H-phosphinic acids proceeds well,
7 it is limited in terms of the ester moiety and is less convenient on small scales. On the other hand, we showed that
H-phosphinic acids can be esterified with alkoxysilanes.
8 Thus, direct treatment of the crude allylation reaction mixtures in DMF with tetrabutoxysilane for 10-16 h at 85 ℃ gave the corresponding butyl esters, which were isolated by chromatography over silica gel in good to excellent yields. Nonetheless, the yields of the allylation/esterification sequence
6 are a little lower than with the direct extraction.
5 In the simplest case of allyl alcohol, a 43% yield of allyl
H-phosphinic acid was isolated after extractive work-up. This is likely due to the more difficult handling and lower hydrolytic stability of the more polar, low molecular weight product. In this case, DBU-promoted alkylation of alkyl phosphinates with allyl bromide was superior.
3gThe 2008 full paper also examined the mechanism of the reaction and the related allylation with allylic acetates, benzoates, and carbonates.
6 It is interesting to note that these substrates performed well, and that DMF could be substituted with CH
3CN.
In this addendum to our original article, we summarize recent extensions in the scope of the methodology, which include replacing hypophosphorous acid with H-phosphinic acids and their esters, using allylic amines or benzylic alcohols instead of allylic alcohols. Some synthetic applications are also included.
Extension to the Synthesis of Disubstituted Phosphinic Acids
The Pd-catalyzed allylation of hypophosphorous acid (H
3PO
2) with allylic alcohols
4,5,6 was subsequently extended to the less reactive
H-phosphinic acids (RPO
2H
2) as shown in Scheme 1.
10 The reactivity of phosphinylidene compounds R
1R
2P(=O)H correlates with the ease of - or rather the less difficult - tautomerization to R
1R
2P-OH, and this can be experimentally determined by measuring the rate of deuteration of R
1R
2P(=O)H into R
1R
2P(=O)D.
10Reactivity/rate of tautomerization increases from electron-donating to less electron-donating, to electron-withdrawing substituents.
11 For example, the half-life of deuteration of H
3PO
3, OctPO
2H
2, PhPO
2H
2, and H
3PO
2, are: 49 h, 5.4 h, 55 min, and 3 min, respectively. Thus, the Pd-catalyzed allylation of
H-phosphinic acids is intrinsically more difficult than of hypophosphorous acid and requires more forcing conditions.
9 First, the reaction solvent and temperature were changed from DMF
4 at 85℃ to
t-AmOH at reflux (102 ℃) in the presence of molecular sieves (3Å, 1 g/mmol) or a Dean-Stark trap. Part of the success of
t-amyl alcohol is attributed to the stabilization of the RP(OH)
2 tautomer via hydrogen-bonding. Second, the catalyst loading required was generally 2 mol% Pd/xantphos (versus 0.5 mol% when H
3PO
2 was the reaction partner). With these modifications, a variety of disubstituted phosphinic acids could be obtained in good to moderate yield after esterification (BnBr/Ag
2O).
9 Again, cinnamyl alcohol proved to be a superb allylating agent.
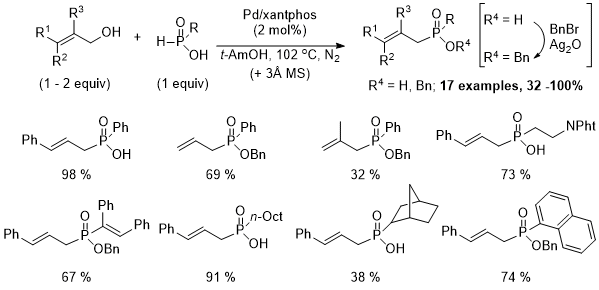
Scheme 1. Palladium-catalyzed allylation of H-phosphinic acids.
Extension to Allylic Amines
In 2014, Tian and coworkers published the analogous allylation of hypophosphorous acid and
H-phosphinic acids with (protonated) allylic amines.
12 As in our reactions, H
3PO
2 is much more reactive than RPO
2H
2, so Pd/xantphos loadings of 0.2 mol% in CH
3CN and 2 mol% in
t-AmOH were used respectively. Scheme 2 shows some of the results.
Scheme 2. Palladium-catalyzed allylation of hypophosphorous acid and H-phosphinic acids with primary allylic amines (22 examples, 68-98 %).
Extension to Benzylation
Having developed the successful allylation of the less reactive
H-phosphinic acids, we then turned our attention to replacing allylic alcohols with benzylic ones. Since the palladium insertion into a benzylic electrophile is significantly more difficult than into an allylic one, more demanding reaction conditions were expected. Higher loadings were necessary as well as higher reaction temperature (Scheme 3).
13Two examples of benzylation of H-phosphinic acid were also provided. Finally, the benzylation of (R)-1-(2-naphthyl)ethanol (97% ee) proceeded in good yield but with significant erosion of the ee to 77%.
Scheme 3. Direct, palladium-catalyzed benzylation of hypophosphorous acid with benzylic and heterobenzylic alcohols.
Extension to H-Phosphinate Esters and Related Compounds
Exactly ten years after the publication of our original reaction,
4 we decided to investigate the reaction of
H-phosphinate esters. Based on mechanistic studies and the resulting postulated mechanism of the allylation/benzylation, esterification of hypophosphorous acid or
H-phosphinic acid is the first step of the transformation. Thus, we did not think that
H-phosphinate esters could give the desired product. However, this assumption was wrong and both allylation and benzylation were accomplished with somewhat narrower scope than those described above.
14 This could be explained by the generally more electron donating nature of the R ester group compared to R=H in the acid, and therefore the ester is less reactive than the acid. Indeed, the half-life of deuteration of
n-OctP(O)(OEt)H, OctPO
2H
2, PhP(O)(OEt)H and PhPO
2H
2, are: 8.2 h, 5.4 h, 1.4 h, and 55 min, respectively.
10 With cinnamyl alcohol, even rather unreactive phosphorus compounds like diethyl
H-phosphonate gave good results (Scheme 4).
14
Scheme 4. Palladium-catalyzed reaction of various phosphorus compounds with cinnamyl alcohol.
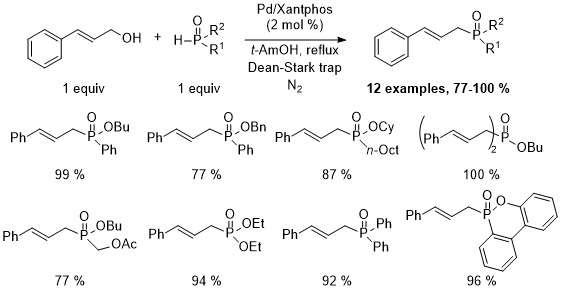
Additional results with different allylic and benzylic alcohols are summarized in Scheme 5.
14 Not shown in Scheme 5 is the reaction between diethyl
H-phosphonate and benzyl alcohol, which gives only 23% of product (
31P-NMR yield). Fortunately, Arbuzov-type reactions have been described to prepare phosphonate diesters from the corresponding benzylic and allylic alcohols.
15,16Scheme 5. Palladium-catalyzed reaction of various H-phosphinate esters with allylic and benzylic alcohols.
Synthetic Applications
If the original conditions are followed by heating in air at 110 ℃, the product can be directly converted into the corresponding phosphonic acid (Scheme 6).
17Scheme 6. One-pot allylation/oxidation preparation of cinnamyl phosphonic acid.
The reaction can also be used to prepare various
P-heterocycles (Scheme 7). In 2008, butyl cinnamyl-
H-phosphinate
1 was allylated through a sila-Arbuzov reaction to
2 or esterified using the Atherton-Todd reaction to produce
3. Both intermediates were cyclized via Grubbs' ring-closing metathesis using catalyst
4, to
P-heterocycles
5 and
6, respectively.
6The same year, the allylation of
H-phosphinic acids became available and symmetrical bis(cinnamyl)phosphinic acid
7 could be synthesized directly in quantitative yield.
9 Silver-promoted esterification gave
8, which was submitted to ring-closing metathesis. Because of the alkene substitution, the reaction required a higher catalyst loading and the yield was lower. From intermediate
8, a different type of heterocycle
10 could be prepared via ozonolysis and double reductive amination.
Later on, once we discovered that
H-phosphinate esters could also be allylated, an improved synthesis of heterocycle
5 became possible (Scheme 7, Montchamp 2016).
14 Monoallylation of hypophosphorous acid to prepare
11 proceeded in excellent yield.
4,5 Because
H-phosphinic acids like
11 can be esterified via azeotropic distillation but disubstituted phosphinic acids like
7 cannot, this allows an inexpensive and efficient access to
1. Allylation of
1 with allylic alcohol produces intermediate
2, this time in a very efficient sequence with only water as a byproduct in each step. Ring closing metathesis forms heterocycle
5 and the yield was improved over the initial cyclization of
2. This streamlined synthesis produces
5 in 4 steps and 70% overall yield.
Scheme 7. Preparation of P-heterocycles using the synthesis of allylic precursors.
In connection with studies aiming at the preparation of aspartate transcarbamoylase (ATCase) inhibitors, ozonolysis of allylated precursors was a key step (Scheme 8). Ozonolysis of
8 as in Scheme 7, but this time using benzylated aspartic acid in the reductive amination step, gave heterocycle
12. Straightforward debenzylation gave
13, which unfortunately showed no inhibition.
18Cinnamyl-
H-phosphinic acid
11 was protected with triethylorthoacetate and the resulting
14 was ozonolyzed and oxidized to carboxylic acid
15. Simple carbodiimide amidation gave
16, which was subsequently deprotected to give
17. Compound
17 is a competitive inhibitor with an inhibition constant of 420 nM, which is approximately 25 times less potent than the known phosphonic acid and anticancer agent PALA.
19Scheme 8. Preparation of potential inhibitors of aspartate transcarbamoylase.
In another application, menthyl (hydroxymethyl)-
H-phosphinate
18 of high diastereoisomeric excess
20 was elaborated into chiral heterocycle
22 (Scheme 9). Dehydrative allylation of
18 under the usual conditions proceeded stereospecifically and gave
19 in nearly quantitative yield. It should be noted that the half-life of deuteration for
18 is remarkably short at only 7 min,
21 thus indicating an unusual reactivity. Reduction of the double-bond to
20 followed by Corey-Kim oxidation delivered menthyl
H-phosphinate
21 in excellent yield and very slight erosion of the diastereoselectivity. Cyclization then gave heterocycle
22, stereospecifically and in excellent yield.
Scheme 9. Preparation of a chiral P-heterocycle.
Additionally, the reaction below has been used for the preparation of a corrosion inhibitor.
22 Palladium-catalyzed allylation of geraniol and oxidation gave geranylphosphonic acid in 60% overall yield (Scheme 10).
Scheme 10. Preparation of geranylphosphonic acid.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved