Ketones are important components of biologically relevant scaffolds and versatile synthetic intermediates.
2 Approaches to synthesize ketones have relied heavily on organometallic reagents, such as the Weinreb ketone synthesis,
3 nucleophilic addition to acid chlorides
4 or aldehydes
5 (a subsequent oxidation step is required for aldehydes), or cross-coupling to activated acyl coupling partners.
6, 7 In the past decade, the synthesis of ketones through cross-electrophile coupling (XEC) has matured, achieving broader functional group tolerance with more commercially available substrate pools. This Addendum discusses recent advancements in XEC approaches to prepare ketones, highlighting new strategies for acyl electrophile activation, practical improvements, and improved mechanistic understanding of these systems. We also note advances in this area have been comprehensively reviewed up to May 2023.
8Ketones from Acyl-X + C(sp3)-X
As is the case with the broader field of XEC, nickel is by far the most commonly used metal catalyst used in this class of reaction. At the time of our initial
Org. Synth. report in 2016 on the synthesis of ketones from carboxylic acid derivatives through XEC,
9 coupling partners in this area were generally limited to acid chlorides with alkyl halides (6 out of 10 reports).
10,11,12,13,14,15 The instability of acid chlorides is a limitation to achieving broad functional group tolerance and we found early success in switching to the more stable 2-pyridyl thioester,
11 inspired by pioneering work from Mukaiyama and co-workers.
16 Concurrently, Hegui Gong and co-workers had showed that carboxylic acid anhydrides could also be converted into the ketone through XEC with alkyl halides.
17,18,19 This approach brought an added benefit that the carboxylic acid could be coupled through an in situ activation to the mixed anhydride. The only example of enantioselective XEC with acyl electrophiles at the time was by Reisman and co-workers, which coupled acid chlorides with secondary benzyl chlorides using a chiral BOX ligand.
13Coupling Partners and Activation Strategies
In the past decade, several other approaches to activate carboxylic acids as acyl electrophiles have been demonstrated in XEC (Figure 1A). Activated esters and thioesters have improved stability relative to acid chlorides, but can readily undergo oxidative addition with nickel. Acid fluorides are also stable alternatives to acid chlorides that can be chromatographed and isolated.
N-Acylimides are part of a class of destabilized amides
20 that have been applied to transition-metal catalyzed cross-coupling, and are beginning to see use in XEC. The emergence of new acyl electrophiles in XEC can often be traced back to acid activation strategies that were originally developed for peptide coupling.
21,22,23 Thus, the most useful acyl electrophiles are those that (1) are isolable and bench stable compounds, (2) can readily undergo oxidative addition with nickel catalysts to form an acylnickel(II) complex, and (3) generate byproducts that do not interfere with the XEC step. There are also less common activation strategies that generate an acyl radical in the mechanism (via the
N-acylimidazole or
N-acylphosphonium). These approaches are distinct from the more common radical decarboxylation in XEC (such as from
N-hydroxyphthalimide esters),
24 which generate an alkyl radical under reducing conditions, rather than an acyl radical.
Concurrently, developments in alkyl electrophile activation have enabled access to the broadest substrate pools in XEC (Figure 1B).
25 Alkyl halides are orders of magnitude more abundant than organometallic reagents and are still the most used alkyl electrophile used to make ketones. Alcohols are most commonly employed as the sulfonate ester, together with a halide salt additive to convert the unreactive sulfonate into the more reactive alkyl halide. Additionally, carboxylic acids (as the
N-hydroxyphthalimide ester) and alkylamines (as the
N-alkylpyridinium salt) can be coupled with acyl electrophiles. Less common types of alkyl coupling partners have also been reported for ketone synthesis such as oxime esters,
26 trimethylammonium salts,
27 and alkyl chlorides.
28Figure 1. Classes of coupling partners in the XEC of acyl electrophiles with alkyl electrophiles and their frequency of use as of July 2025
There have been increased efforts to generate acyl electrophiles in situ from the acid to reduce step count and increase synthetic utility (Figure 2). Key considerations for these strategies are (1) clean and complete conversion to the activated electrophile in a reasonable reaction time, (2) solvent compatibility for both the activation step and the XEC step in order to avoid a solvent swap, and (3) the XEC step must be able to tolerate the byproducts generated from the activation step. Hegui Gong's pioneering reports on acid anhydride couplings largely remains the most common method for in situ activation of carboxylic acids in XEC.
17,18,19 Typical conditions use an excess of di-
tert-butyl dicarbonate (Boc anhydride). Acid fluorides can be generated in situ from tetramethylfluoroformamidinium hexafluorophosphate (TFFH) and Proton Sponge as base.
29 Recently, we showed that di-2-pyridyl carbonate (DPC) with catalytic 4-dimethylaminopyridine (DMAP) can be used to generate 2-pyridyl esters in situ for subsequent XEC.
30
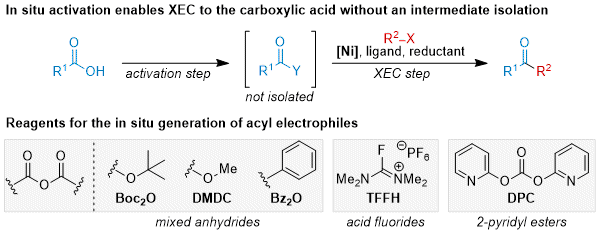
Figure 2. Reagents for the in situ generation of acyl electrophiles from carboxylic acids
Mechanism
In addition to substrate pool analysis, a mechanistic understanding of XEC is key to the development of new methodology in this area. While the details of each mechanistic step can change based on conditions, the general mechanism for C(sp
2)-C(sp
3) XEC is shown in Figure 3.
31 In this paradigm, a C(sp
2) acyl electrophile (isolated or generated in situ) undergoes oxidative addition with nickel through a non-radical mechanism, generating an acylnickel(II) complex. Depending on reaction conditions and alkyl radical precursor, a variety of mechanisms can generate the C(sp
3) radical, which reacts with the acylnickel(II) species to generate a nickel(III) intermediate. This readily undergoes reductive elimination to afford the ketone and a nickel(I) species. Reduction of the nickel catalyst turns over the catalytic cycle. The most common reductants are Zn and Mn, but photochemical
32,33,34,35,36,37 and electrochemical
38,39,40 strategies are increasingly employed.
While a detailed discussion of XEC mechanism is outside the scope of this Addendum, there is an alternative route specific to acyl electrophiles involving a reversible decarbonylation step.
17, 18, 41, 42 We
43, 44 and others
45,46,47 have shown that a CO migration and extrusion step can precede radical capture and lead to an arylnickel(II) or alkylnickel(II) under certain conditions. Subsequent radical capture at this intermediate and reductive elimination generates the R-C(sp
3) product. This mechanism can be favored when (1) temperature is elevated (often >100 ℃), (2) selection of the acid activating group can promote the decarbonylation equilibrium, or (3) CO can escape the system (e.g., by an N
2 flush), which prevents any reversibility.
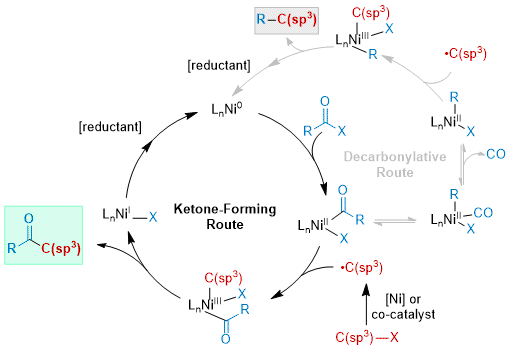
Figure 3. General mechanism for the C(sp2)-C(sp3) cross-electrophile coupling of acyl electrophiles with alkyl electrophiles. While the ketone route generally predominates, under certain conditions the decarbonylative route can become the major product-forming mechanism
New Modes of Alkyl Activation Strategies Applied to Ketone Synthesis
A notable advancement in XEC is the development of new approaches to couple substrate pools outside of alkyl halides. The emergence of N-hydroxyphthalimide (NHP) esters and N-alkylpyridiniums has led to increased interest in developing improved conditions to prepare ketones. Indeed, 79% of new reactions for C(sp2)-C(sp3) XEC for ketone synthesis were reported after 2016.
In 2019, our group
48 and Baran
49 concurrently developed conditions to cross-couple two carboxylic acids for ketone synthesis (Figure 4). This type of strategy requires distinct activation modes for each carboxylic acid coupling partner and that they do not scramble under reaction conditions. Our report, in collaboration with Gellman, employed 2-pyridyl thioesters (as the acyl donor) and alkyl NHP esters (as the radical donor). This report also showed how NHP ester consumption could be tuned by the addition of zinc salts, and by using a mixed solvent system of THF and DMA. Baran's report had a similarly broad scope using in situ generated mixed anhydrides as the acyl donor. Later reports have enabled this type of coupling to be driven photochemically with Hantzsch ester as reductant.
33,34,35,36,37Figure 4. Ketones from coupling two carboxylic acids
Amines are also an abundant substrate pool and most commonly associated with C-N amide bond formation when reacted with a carboxylic acid. The advancement of
N-alkylpyridinium salts as radical sources has enabled the use of these coupling partners in C-C bond formation. These reagents commonly prepared by the condensation of a primary or secondary amine with commercially available 2,4,6-triphenylpyrillium tetrafluoroborate. Soon after the emergence of these coupling partners in cross-coupling with aryl boronic acids,
50 reports of XEC began to appear (Figure 5). The Matsuo group showed that
N-aroylsuccinmides could be coupled to
N-alkylpyridinium salts.
51 Rasappan and co-workers performed this coupling with acyl chlorides or in situ generated anhydrides.
52 Our group, in collaboration with the Watson group, showed that in situ generated acid fluorides could be coupled to
N-alkylpyridiniums, including several examples of complex amine fragments derived from natural products.
29 In the same report we also developed modified conditions for secondary
N-alkylpyridiniums, swapping the acyl electrophile to a 2-pyridyl ester and the ligand to Bphen. Later, Kranthikumar and co-workers reported a ketone synthesis from the XEC of 2-pyridyl thioesters with
N-alkylpyridiniums.
53
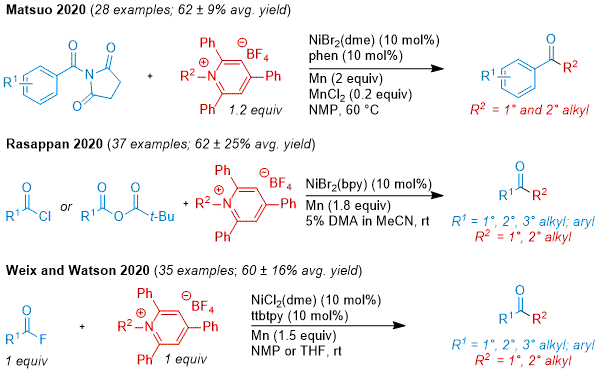
Figure 5. Ketones from coupling a carboxylic acid with an amine
Alkyl alcohols represent another important substrate pool, and at time of writing they have been most generally applied to ketone synthesis as the sulfonate ester.
15, 54, 55 In these reactions, in situ halide exchange introduced by a salt additive or the nickel precatalyst generates low concentrations of the alkyl halide, which is the active coupling partner. Deoxygenative XEC is far less developed in ketone synthesis despite their rapid advancement in other areas of XEC (e.g., aryl-alkyl bond formation). This is perhaps due, in part, to the challenge of overcoming transesterification of the free alcohol with the acyl electrophile. A recent report from Fleischer and co-workers disclosed promising conditions to couple thiophenyl esters with benzyl alcohols through nickel/titanium co-catalysis, although the scope is limited to methoxy-substituted benzyl alcohols.
56Stereochemical Control
Since the Reisman group's pioneering work on the enantioconvergent XEC of acid chlorides with secondary benzyl chlorides,
13 new approaches to make α-chiral ketones have focused on coupling other classes of activated alkyl electrophiles (Figure 6). The groups of Xi-Sheng Wang, Genping Huang & Chun Zhang, and Liang-An Chen all reported conditions to couple acid chlorides with α-trifluoromethyl alkyl bromides.
57,58,59 Shaolin Zhu and co-workers demonstrated a two-ligand system based on their previous work
60 that enabled a migratory, enantioconvergent coupling of alkyl iodides with carboxylic acids (via in situ anhydride generation), synthesizing chiral α-arylated ketones via chain-walking.
61 Liang-An Chen, Qiaorong Han, and co-workers successfully coupled acid chlorides to α-bromobenzoates to synthesize chiral acyloin products.
62 Baran and co-workers reported the coupling of acid chlorides with amino acid-derived NHP esters to prepare chiral α-amino ketones.
63 Reisman, Sigman and co-workers reported the synthesis of α-chiral ketones through the desymmetrization of cyclic
meso-anhydrides with benzyl chlorides.
46 This report also disclosed conditions for the decarbonylative XEC of
meso-anhydrides with primary alkyl bromides and a wealth of mechanism-driven ligand design. In their studies of enantioconvergent XEC to 2-aryloxetanes via ring-opening, Kaiwu Dong and co-workers reported conditions that could couple to acid anhydrides.
64 Across these reports, it is worth noting the structural similarity of the BOX ligands for these transformations. In analogy to other types of enantioselective C(sp
2)-C(sp
3) XEC, this area has seen the broadest success with activated alkyl electrophiles, the majority of examples being with alkyl halides. Mechanistically, enantioconvergent couplings remain most common, as XEC with alkyl electrophiles proceeds through an alkyl radical.
In a different class of stereochemical challenge, we reported an approach to prepare (
E)-enones from coupling acrylic acids (as the in situ generated 2-pyridyl ester) with alkyl bromides.
30 In this case, stereochemical control comes from thermodynamic preferences accessed by nickel-mediated
E/Z isomerization as the reaction progresses.
Figure 6. Recent advances in the enantioselective XEC of acyl electrophiles
Applications to Synthesis
The improved functional group tolerance offered by XEC over alkyl nucleophiles in cross-coupling can prove beneficial in assembling complex fragments for the synthesis of natural products. The Kishi group has reported a series of XEC reactions to prepare dialkyl ketones that have been applied to total synthesis (Figure 7). The initial report employed an iron-catalyzed, copper-mediated XEC of acyl electrophiles with alkyl iodides.
65 Soon after, their group reported a nickel-catalyzed system where a zirconium additive is key to activate the alkyl iodide reduction.
66 Improved conditions were later reported that enabled a 1:1 stoichiometry of 2-pyridyl thioester to alkyl iodide with significantly lower catalyst loading.
67 These conditions tolerate sensitive functionality such as C(sp
2)-halides, alcohols, and phenols. Kishi applied these conditions to assemble late-stage fragments in syntheses of the halichondrin
67,68,69 and halistatin
70 classes of natural products. In a joint research effort between scientists at Eisai and the Kishi group, this chemistry was scaled to deliver over ten grams of E7130, an anticancer clinical candidate and a structural analogue of halichondrin B.
71, 72Others molecules that have been completed using the Ni/Zr XEC system include (-)-irijimaside A by Umehara and Sasaki,
73 and bryostatins 1, 7, and 9 by Zhenlei Song and co-workers.
74 Additionally, Chulbom Lee and co-workers successfully coupled a pyridyl thioester with an NHP ester using modified conditions of the Ni/Zr system for the synthesis of a fragment of madeirolide A.
75 Outside of the Ni/Zr approach, Shuanglin Qu, Qianghui Zhou, and co-workers reported a synthesis of (-)-berkelic acid by coupling an α-tertiary acyl chloride with a primary alkyl iodide.
76Ketones from Acyl-X + C(sp2)-X
The XEC of an acyl electrophile with other C(sp2) electrophiles has been slower to develop than C(sp2)-C(sp3) acyl-alkyl coupling. Cross-selectivity in these systems can be difficult to control as now each coupling partner reacts with nickel through a two-electron oxidative addition mechanism. This results in unselective statistical mixtures if the coupling partners react at similar rates or homodimerization if they aren't inherently well-matched. Representative examples are shown in Figure 8.
Figure 7. Application of nickel/zirconium XEC to prepare halichondrins and analogous natural products
Jianlin Han and co-workers demonstrated the feasibility C(sp
2)-C(sp
2) acyl coupling through the XEC of
N-acylglutarimides with aryl iodides to prepare diaryl ketones.
77 Walsh, Jianyou Mao, and co-workers reported the desymmetrizing XEC of cyclic
meso-anhydrides with aryl triflates.
78 Xing-Zhong Shu and co-workers developed a synthesis of enones through the XEC of aroyl fluorides with cyclic vinyl triflates.
79 An alternative strategy to achieve cross-selectivity is the selection of conditions that proceed through an acyl radical to differentiate its reactivity from the other C(sp
2) coupling partner. In their study on the XEC of
N-acylimidazoles, which generates an acyl radical for aroyl substrates, Chao Li and co-workers showed several examples coupling to aryl bromides.
80 Jia Xie ad co-workers reported a Ni/Ir photocatalytic system where triphenylphosphine can be used to generate an acyl radical via an acylphosphonium.
81 This has translated to successful couplings of benzoic acid derivatives with aryl bromides (to prepare diaryl ketones)
82 and vinyl triflates (to prepare enones).
83
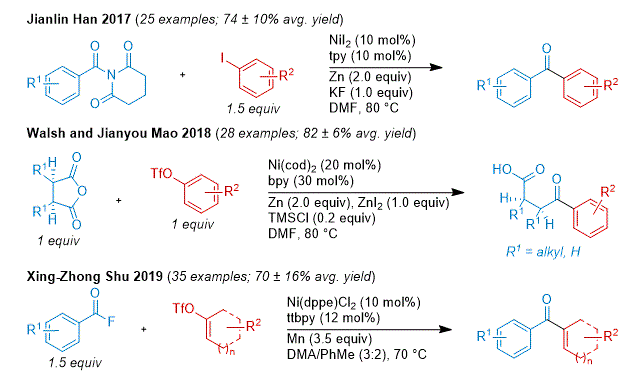
Figure 8. Representative examples of ketone synthesis from the XEC of acyl electrophiles with C(sp2) electrophiles
Despite these advancements, XEC approaches to prepare diaryl ketones remains underdeveloped, relying more on nucleophiles (e.g., Suzuki coupling of aroyl electrophiles with aryl boronic acids) to achieve cross-selectivity. Strategies used to distinguish C(sp2) electrophiles in biaryl XEC such as multimetallic couplings have yet to be reported with an acyl electrophile. The scope of acyl-X + C(sp2)-X couplings is also notably limited compared to other classes of XEC, with no examples of aromatic nitrogen heterocycles reported to date.
The synthesis of ketones by XEC has rapidly grown since the time of our Org. Synth. report in 2016. The development of new strategies to activate carboxylic acids as electrophiles has been a significant contributor to this interest. 2-Pyridyl (thio)esters are especially useful as they have increased stability relative to acid chlorides but still readily undergo oxidative addition with nickel. Mixed anhydrides remain the most common approach for in situ activation. The XEC of acyl electrophiles with C(sp3) electrophiles has seen the largest number of reports, in analogy to the rapid growth of C(sp2)-C(sp3) aryl-alkyl XEC. However, as is the case with the entire field, achieving robust cross-selectivity can be challenging, as is evident in the coupling acyl electrophiles with other C(sp2) electrophiles. The XEC with acyl electrophiles relies on a relatively small set of nitrogen-based ligands (bipyridines, terpyridines, and phenanthrolines), and the development of new ligands could promote new reactivity. Certain classes of coupling partners remain challenging for XEC with acyl electrophiles, such acrylic acid-derived C(sp2) electrophiles and deoxygenative couplings of C(sp3) alkyl alcohols. Enantioselective syntheses have also developed to include other classes of activated electrophiles through enantioconvergent couplings, and there will undoubtedly continue to be advancements in this rapidly growing area.
Recently, there has been increased interest in controlling the equilibrium arising from loss of CO from acylnickel(II) intermediates to promote the decarbonylative coupling and synthesize aryl-alkyl and alkyl-alkyl bonds from carboxylic acids. This provides complementary reactivity and products to the corresponding acyl coupling. As the field's understanding of XEC mechanism, ligand design, and substrate pool activation modes continue to improve, we expect the XEC of acyl electrophiles will continue to be one of the most widely used approaches to synthesize ketones.
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved