Org. Synth. 2005, 81, 33
DOI: 10.15227/orgsyn.081.0033
4-NONYLBENZOIC ACID
[(Benzoic acid, 4-nonyl-)]
Submitted by Alois Fürstner
1, Andreas Leitner, and Günter Seidel.
Checked by Renee Kontnik and Steven Wolff.
1. Procedure
A. 4-Nonylbenzoic acid methyl ester. A 250-ml, three-necked, round-bottomed flask equipped with a Teflon-coated magnetic stirbar, glass stopper, a reflux condenser fitted with an argon stopcock inlet, and a pressure-equalizing dropping funnel is charged with magnesium turnings (2.95 g, 121.0 mmol). The flask is evacuated and flame-dried while the magnesium turnings are gently stirred. After reaching ambient temperature, the apparatus is flushed with argon, the magnesium turnings are suspended in 20 mL of tetrahydrofuan (Note 1), and 1,2-dibromoethane (0.3 ml, 3.6 mmol) (Note 2) is introduced. A solution of 1-bromononane (20.52 g, 97.0 mmol) (Note 2) in 100 mL of THF (Note 1) is then added via the addition funnel to the suspension over a period of ca. 45 min at such a rate as to maintain gentle reflux. After the addition is complete, the mixture is refluxed for another 20 min and then the resulting solution of nonylmagnesium bromide is allowed to cool to ambient temperature. In the meantime, an oven-dried, 2-L, two-necked, round-bottomed flask equipped with a magnetic stirbar, rubber septum, and an argon inlet is flushed with argon and charged with 4-chlorobenzoic acid methyl ester (13.0 g, 76.2 mmol) (Note 2), ferric acetylacetonate [Fe(acac)3] (1.35 g, 3.82 mmol) (Note 2), 450 ml of THF (Note 1), and 25 mL of N-methylpyrrolidinone (NMP) (Note 1). The flask is immersed in an ice bath and the solution of nonylmagnesium bromide prepared above is immediately added within one min via a polyethylene cannula. This causes an immediate color change from red to black-violet. The ice bath is removed and the resulting dark mixture is stirred for 7-10 min at ambient temperature, diluted with 200 mL of diethyl ether, and then carefully quenched by addition of 300 mL of 1M HCl with stirring. The mixture is transferred into a separatory funnel (the flask is rinsed with 200 mL of Et2O), and the aqueous phase is separated and extracted with 200 mL of Et2O. The combined organic phases are washed with 300 mL of saturated aq NaHCO3, dried over Na2SO4, and concentrated by rotary evaporation at reduced pressure (Note 3). The resulting crude orange-red residue is purified by short path distillation under high vacuum (1 × 10−4 torr) to give 15.81-16.85 g (79-84%) of 4-nonylbenzoic acid methyl ester as a colorless syrup, bp 103-105°C (Notes 4, 5).
B. 4-Nonylbenzoic acid. A 500-mL, round-bottomed flask equipped with a Teflon-coated magnetic stirbar and a reflux condenser is charged with 4-nonylbenzoic acid methyl ester (10.07 g, 38.37 mmol), 100 mL of methanol (Note 1), and 96 mL of 1M aqueous NaOH. The resulting mixture is heated at reflux for 18 hr and then allowed to cool to room temperature. The reaction mixture is carefully acidified by addition of 200 mL of 1M aqueous HCl, and the resulting solution is transferred to a separatory funnel and extracted with four 250-mL portions of ethyl acetate. The combined organic layers are dried over Na2SO4, filtered, and concentrated by rotary evaporation at reduced pressure. The residue (ca. 9.5 g) is recrystallized from 70 mL of hexanes to give 8.32-8.35 g (87-88%) of 4-nonylbenzoic acid as a white solid (Notes 6, 7).
2. Notes
1.
The submitters used THF that was freshly distilled over Na/K alloy; NMP was distilled over CaH
2. The checkers used commercially available anhydrous
THF and NMP obtained from Aldrich Chemical Co. There appears to be a slight increase in yield when
THF distilled from sodium/benzophenone ketyl is used instead of the commercial THF. All other solvents used were of reagent grade quality and were used without further purification.
2.
1,2-Dibromoethane, 1-bromononane (98%), 4-chlorobenzoic acid methyl ester (99%), and Fe(acac)3 (99.9%) were purchased from Aldrich Chemical Co. and used as received.
3.
The checkers found that a dark red oil separated upon concentration of the ethereal solution of the crude ester. In this instance, the ester was diluted with
200 mL of ether, washed with
200 mL of brine, and then again concentrated.
4.
The submitters obtained the product in
87-90% yield.
5.
The product is ≥ 95% pure by GC (Agilent 6890 Series, column: HP-5MS; 5% phenyl methyl siloxane, 30 m × 250 (m 0.25 (m); the remainder is octadecane formed by oxidative coupling of the Grignard reagent; T-program: 70°C (3.5 min)→280°C (20°C/min); 12.62 min retention time;
1H NMR (300 MHz, CDCl
3): δ 0.86 (t, J = 6.7 Hz, 3H), 1.2-1.36 (m, 12H), 1.60 (m, 2H), 2.63 (t, J = 7.7 Hz, 2H), 3.88 (s, 3H), 7.22 (d, J = 8.4 Hz, 2H), 7.93 (d, J = 8.3 Hz, 2H);
13C NMR (75 MHz, CDCl
3): δ 14.1, 22.7, 29.2, 29.3, 29.4, 29.7, 31.1, 31.9, 36.0, 51.9, 127.6, 128.4, 129.6, 148.5, 167.2; IR (film): 3032, 2953, 2926, 2855, 1725, 1611, 1465, 1435, 1278, 1178, 1109, 1021, 854 cm
−1; MS (El):
m/z (rel. Intensity) 262 (63, [M
+]), 231 (24), 163 (13), 150 (100), 121 (8), 105 (9), 91 (41), 57 (8), 43 (13). Anal. Calcd for C
17H
26O
2: C, 77.82; H, 9.99. Found C, 78.30; H, 10.45.
6.
The submitters obtained the product in
94% yield.
7.
The product has the following physical properties:
mp 92.5-94.3°C;
1H NMR
pdf(400 MHz, CDCl
3): δ 0.86 (t, J = 6.9 Hz, 3H), 1.2-1.38 (m, 12H), 1.62 (m, 2H), 2.66 (t, J = 7.7 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 8.01 (d, J = 8.2 Hz, 2H);
13C NMR (100 MHz, CDCl
3): δ 14.1, 22.7, 29.2, 29.3, 29.4, 29.5, 31.1, 31.8, 36.1, 126.7, 128.6, 130.4, 149.6, 171.9; IR (film): 3072, 2924, 2852, 2669, 2554, 1683, 1609, 1575, 1469, 1424, 1321, 1290, 945, 859, 758 cm
−1; MS (El):
m/z (rel. intensity): 248 (56, [M
+]), 177 (7), 149 (9), 136 (100), 107 (9), 92 (38), 57 (14); 29 Anal. Calcd for C
16H
24O
2-H
2O: C, 72.14; H, 9.84. Found C, 72.53; H, 9.76.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Modern cross coupling chemistry is heavily dominated by the use of palladium and nickel complexes as the catalysts, which show an impressively wide scope and an excellent compatibility with many functional groups.
2 This favorable application profile usually overcompensates the disadvantages resulting from the high price of the palladium precursors, the concerns about the toxicity of nickel salts, the need for ancillary ligands to render the complexes sufficiently active and stable, and the extended reaction times that are necessary in certain cases.
Our group has recently developed an alternative method for alkyl-(hetero)aryl- as well as aryl-heteroaryl cross coupling reactions catalyzed by iron salts.
3,4 This methodology was inspired by early reports of Kochi et al.
5,6 on iron-catalyzed cross coupling of vinyl halides and is distinguished by several notable advantages.
- The expensive noble metal catalysts are replaced by cheap, air stable, commercially available and toxicologically benign iron salts without any loss in efficiency. The reactions are usually carried out under "ligand free" conditions using inexpensive Grignard reagents as the preferred coupling partners.
- Aryl chlorides, which are the most attractive type of substrates due to their low cost and ready availability, perform inherently better than the corresponding aryl bromides or iodides. Moreover, aryl triflates and even electron-deficient aryl tosylates can be used as the starting materials.
- Most iron-catalyzed reactions proceed at unprecedentedly high rates and are finished within a few minutes even when carried out at or below ambient temperature.
- Due to the efficiency with which the iron catalysts activate the C-Cl bond, several functional groups are tolerated that normally would react with a Grignard reagent.
- When applied to polyfunctional substrates, iron catalyzed reactions allow either for selective, exhaustive, or consecutive cross-coupling processes to be carried out in "one pot."
Most of these chemical features are evident from the preparation of
4-nonylbenzoic acid described above which requires less than 10 min reaction time at 0°C→ r.t. even when performed on a > 15 g scale. No competing attack of the Grignard reagent onto the methyl ester of the substrate can be detected under the reaction conditions. It should be noticed that
4-nonylbenzoic acid, like other alkyl benzoic acid derivatives that are equally available by this iron-catalyzed cross coupling method, is of some practical relevance as liquid crystalline material or a component thereof.
7,8 The additional examples compiled in the Table illustrate the scope and performance of this new protocol in more detail.
3,4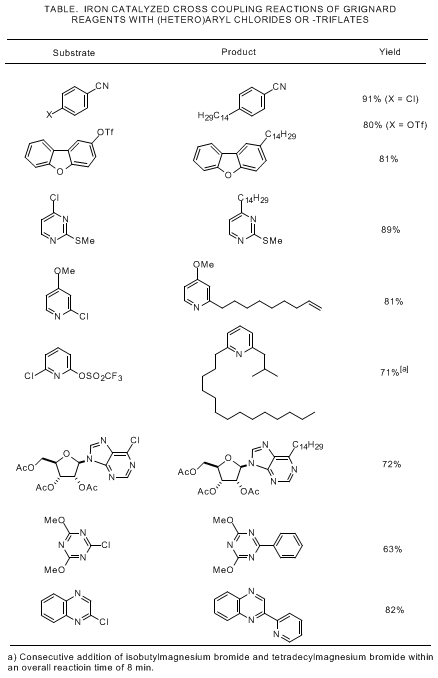
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
4-Nonylbenzoic Acid: Benzoic acid, 4-nonyl-; (38289-46-2)
Magnesium; (7439-95-4)
1-Bromononane:
Nonane, 1-bromo-; (693-58-3)
Nonylmagnesium bromide:
Magnesium, bromononyl-; (39691-62-8)
Ferric acetylacetonate:
Tris (2,4-pentanedionato)iron (III); (14024-18-1)
N-Methylpyrrolidinone:
2-Pyrrolidinone, 1-methyl-; (872-50-4)
4-Chlorobenzoic acid methyl ester:
Benzoic acid, 4-chloro-, methyl ester; (1126-46-1).
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved