Org. Synth. 1985, 63, 66
DOI: 10.15227/orgsyn.063.0066
ENANTIOSELECTIVE EPOXIDATION OF ALLYLIC ALCOHOLS: (2S,3S)-3-PROPYLOXIRANEMETHANOL
[Oxiranemethanol, 3-Propyl-, (2S,3S)-]
Submitted by J. Gordon Hill
1, K. Barry Sharpless
1, Christopher M. Exon
2, and Ronald Regenye
2.
Checked by Mark H. Norman and Clayton H. Heathcock.
1. Procedure
Caution! Reactions and subsequent operations involving peracids and peroxy compounds should be run behind a safety shield. Peroxy compounds should be added to the organic material, never the reverse. New or unfamiliar reactions, particularly those run at elevated temperatures, should be run first on a small scale. Highly concentrated solutions of TBHP are potentially hazardous and can undergo violent decomposition upon exposure to certain metal salts. Strong acids must never be added to high-strength TBHP solutions. Highly concentrated solutions of TBHP are best stored in high-density polyethylene bottles rather than glass because of the potential for pressure buildup due to decomposition forming oxygen gas. For additional discussion, see Gao, Y.; Hanson, R. M.; Klunder, J. M.; Ko, S. Y.; Masamune, H.; Sharpless, K. B. J. Am. Chem. Soc. 1987, 109, 5765-5780. [Note added November 2011].
A 2-L, three-necked, round-bottomed flask equipped with a mechanical stirrer with Teflon blades, thermometer, and nitrogen inlet is charged with 1.00 L of methylene chloride (Note 1) and 39.9 mL (38.1 g, 0.134 mol) of titanium(IV) isopropoxide (Note 2). The flask content is stirred and cooled under nitrogen in a dry ice–ethanol bath to −70°C. To the flask is then added 33.1 g (27.5 mL, 0.161 mol) of diethyl (2R,3R)-tartrate (Note 3) and 25.0 g (0.25 mol) of E-2-hexen-1-ol (Note 4). A small volume of methylene chloride is used to ensure complete transfer of each material to the reaction flask. To the flask is then added 184.5 mL (0.50 mol) of 2.71 M anhydrous tert-butyl hydroperoxide in toluene (Note 5) that has been precooled to −20°C (Note 6). The addition causes a temperature increase to −60°C; the temperature of the reaction mixture is allowed to come to 0°C over a 2.0-hr period (Note 7).
A 4-L beaker equipped with a magnetic stirring bar and thermometer is charged with a solution of 125 g of ferrous sulfate and 50 g of tartaric acid in a total volume of 500 mL of deionized water. The solution is stirred and cooled by means of an ice–water bath to 10°C. When the epoxidation reaction mixture reaches 0°C, it is immediately (Note 8) poured into the stirred contents of the beaker. The resulting reaction is mildly exothermic, causing a temperature rise to ca. 20°C (Note 9). After the exothermic reaction has subsided and the temperature has begun to drop (ca. 5 min), the cooling bath is removed and the mixture is stirred at ambient temperature for 30 min. The contents of the beaker are transferred to a 2-L separatory funnel and the aqueous phase is separated and extracted with two 250-mL portions of ether. The combined organic layers are dried over sodium sulfate and filtered. The solvent is removed with a rotary evaporator at 35°C (70 mm) to give 85.9–89.9 g (Note 10) of pale-amber oil.
A 2-L, three-necked, round-bottomed flask equipped with a thermometer and a mechanical stirrer with Teflon blades is charged with a solution of the reaction product in 750 mL of ether. The contents of the flask are cooled in an ice–water bath to 3°C. To the flask is added a precooled (3°C) solution of 20 g (0.50 mol) of sodium hydroxide in 500 mL of brine (Note 11). The two-phase mixture is stirred vigorously for 1 hr with continued cooling (Note 12) and then is transferred to a separatory funnel. The aqueous phase is separated and extracted with two 150-mL portions of ether (Note 13) and (Note 14). The combined organic solution (Note 15) is dried over sodium sulfate and filtered. Solvent removal with a rotary evaporator at 35°C (70 mm) followed by concentration with the rotary evaporator at 35°C (12 mm) for 1.0 hr gives 24.7–25.0 g of crude (2S,3S)-3-propyloxiranemethanol as a pale-amber oil (Note 16).
The crude product is distilled through a
10-cm Vigreux column (the receiving flask is cooled in an ice–water bath) to yield
22.45–22.84 g (
80–81%) of
(2S,3S)-3-propyloxiranemethanol as a colorless liquid, bp
31–33°C (0.30–0.40 mm). Analysis by GC indicates a chemical purity of 89–93%
(Note 17). The material is fractionally distilled through a
20-cm vacuum-jacketed Vigreux column to obtain
17.69–19.44 g (
63–69%),
[α]D22 −38.1 to −38.6° (neat),
[α]D23 −46.2 to −48.6° (CHCl
3,
c 1.0) of a colorless liquid. Analysis by GC indicates a chemical purity of 96–98%
(Note 17). An enantiomeric purity of 96.4–97.5% is determined by
1H NMR analysis of the derived acetate using
Eu(hfc)3, tris[3-(heptafluoropropylhydroxymethylene)-d-camphorato] europium(III),
(Note 18) as the chiral shift reagent
(Note 19). The enantiomeric purity may also be determined by GC analysis
(Note 20) of the derived
α-methoxy-α-(trifluoromethyl)phenylacetic acid esters3 (Note 21) and
(Note 22). An alternative to the distillation method is purification by preparative HPLC
(Note 23) and
(Note 24) and bulb-to-bulb distillation
(Note 25) to give
21.85 g (
78%) of
(2S,3S)-3-propyloxiranemethanol as a white solid, mp
19°C, [α]
D25 −46.6° (CHCl
3,
c 1.0). Analysis by GC of material purified in this manner indicates a chemical purity of >99%
(Note 14) and an enantiomeric purity of 96.8%
(Note 20),
(Note 21),
(Note 22).
2. Notes
1.
Fisher Scientific Company methylene chloride, certified ACS grade containing 0.02% water was used; a fresh bottle was used for each run.
2.
Titanium(IV) isopropoxide is available from the Aldrich Chemical Company, Inc.3.
(+)-Diethyl L-tartrate was obtained from the Aldrich Chemical Company, Inc.4.
trans-2-Hexen-1-ol was obtained from Alfa Products, Morton Thiokol, Inc. Analysis by GC (Hewlett-Packard HP 5710A; 50-m ×
0.25-mm capillary column of bonded CPS-2 on fused silica; 120°C isothermal) with appropriate standards indicated an
E-2-hexen-1-ol content of 96.1% with 0.9% Z-2-hexen-1-ol and 2.9% hexanol as impurities.
trans-2-Hexen-1-ol from the Aldrich Chemical Company, Inc., could also be used. This material was of similar composition:
95.4% E-2-hexen-1-ol, 0.8% Z-2-hexen-1-ol and 3.2% hexanol.
5.
Anhydrous
tert-butyl hydroperoxide in
toluene4 was prepared starting with
Aldrich Chemical Company, Inc. 70% aqueous tert-butyl hydroperoxide. A 500-g lot of this material was swirled in a separatory funnel with 1.0 L of
toluene. The aqueous phase was removed and discarded. The organic solution was heated at reflux under
nitrogen for 4 hr in a flask equipped with a Dean–Stark trap for water separation (for greater detail, see
4). The solution was cooled and stored under
nitrogen at −20°C
(Note 6). The content of
tert-butyl hydroperoxide was determined by
1H NMR according to the equation
Molarity =
X / 0.1
X + 0.32
Y
where
X = integration of
tert-butyl resonance;
Y = integration of methyl resonance.
6.
Storage of the solution at −20°C is not necessary.
4 It does, however, provide a convenient method of precooling the material.
7.
Addition of dry ice to the cooling bath was stopped. If the rate of temperature increase was too slow, the ethanol bath was lowered.
8.
If the mixture is allowed to stand at 0°C or to warm above this temperature, undesired by-products (TLC) are formed.
9.
The temperature should be kept ≤20°C. In some cases, the addition of small amounts of ice to the reaction is necessary.
10.
This weight can vary substantially, depending on the extent to which the solvents, especially
toluene, have been stripped from the solution. Concentration need only be carried out until the weight is < 100 g.
11.
This mixture is prepared by dissolving the
sodium hydroxide in the
brine at ambient temperature and then cooling the total to 3°C. The resultant cloudy, supersaturated suspension is used in toto. The use of this reagent ensures complete extraction (vide infra) of the somewhat water-soluble product. In addition, it minimizes contact of this material with the aqueous base, conditions which can lead to the Payne rearrangement.
512.
Saponification serves to remove the
diethyl tartrate as well as to liberate any product which has been transesterified to form a tartrate ester.
13.
GC analysis
(Note 14) of a third extract showed no product.
14.
GC analysis was performed on a column with the following properties: Hewlett-Packard HP 5702A,
2-m × 0.63-cm OV-101 column, programmed 70–200°C at 8°C/min.
15.
GC analysis
(Note 14) shows no
diethyl (2R,3R)-tartrate, indicating that the saponification was complete.
16.
Material of this quality is suitable for many synthetic purposes.
17.
The properties of the column are as follows: Hewlett-Packard HP 5790A, 12-m ×
0.2-mm cross-linked methyl silicone (fast analysis) column, programmed from 35 to 140°C at 3°C/min.
18.
The shift reagent was obtained from the Aldrich Chemical Company, Inc. Drying the Eu(
hfc)
3 overnight with a drying pistol at 56°C (refluxing acetone) under vacuum afforded optimum results.
19.
The analytical sample of the acetate derivative is prepared as follows. Into a
5-mL, round-bottomed flask equipped with a magnetic stirring bar are placed 2 drops of the reaction product, 16 drops of
acetic anhydride, and 32 drops of
pyridine. The solution is stirred at ambient temperature for 2 hr, and the mixture is then transferred to a separatory funnel with the aid of
10 mL of methylene chloride. The
methylene chloride solution is washed with
two 10-mL portions of 1 M phosphoric acid, the organic layer is dried over MgSO
4, and the filtered solution is concentrated with a rotary evaporator to give approximately 20 mg of acetate as a colorless oil. A 5-μL sample of this crude acetate is dissolved in
0.5 mL of benzene-d6 and transferred to an
NMR tube. A solution of
75 mg of Eu(hfc)3 in 0.5 mL of benzene-d6 is prepared. A 50-μL portion of the shift reagent solution is added to the acetate sample, the mixture is shaken well, and the
1H NMR spectrum is recorded. Additional portions of shift reagent are added in 10-μL portions until the acetate methyl resonance (originally at δ = 1.65 ppm) shifts downfield to the region 2.3–3.1 ppm and shows baseline resolution of the resonances from the two enantiomers. A total of 50–90 μL of the shift reagent solution should be required to achieve the desired shift, at which point a chemical-shift difference of about 0.2 ppm should be obtained. The %ee is obtained by integration of the two acetate peaks.
20.
The column had the following properties: Hewlett-Packard HP 5710A, 50-m ×
0.25-mm capillary column of OV-17 (bonded) on fused silica; 175°C isothermal.
21.
The analytical sample of
α-methoxy-α-(trifluoromethyl)phenylacetic acid ester is prepared as follows. Into a
5-mL, capped, amber vial equipped with a magnetic stirring bar are placed 20 mg of the reaction product,
1.0 mL of methylene chloride, 87 mg of (+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (Note 22), 4 drops of triethylamine, and 1 crystal of 4-dimethylaminopyridine. The mixture is stirred at ambient temperature for 1.5 hr, at which point TLC
(Note 26) indicates complete conversion to the ester. Addition of
4 drops of N,N-dimethyl-1,3-propanediamine and concentration on a rotary evaporator at 35°C (70 mm) affords a yellow oil. This material is filtered through 10 g of E. Merck silica gel 60 (70–230 mesh) with
9 : 1 hexanes–ethyl acetate until TLC analysis indicates no further product elution. The total eluate is concentrated on a rotary evaporator at 35°C (70 mm). The resultant colorless oil is subjected to GC analysis. A sample of
E-2-hexen-1-ol in
methylene chloride was epoxidized at 20°C with
m-chloroperoxybenzoic acid. The resultant racemic
epoxy alcohol, on conversion to the diastereomeric
(+)-α-methoxy-α-(trifluoromethyl) phenylacetic acid esters in the manner described above, provided a GC standard for determination of the enantiomeric excess obtained in the asymmetric epoxidation.
22.
The acid chloride was prepared
3 from
(+)-α-methoxy-α-(trifluoromethyl)phenylacetic acid, which was used as obtained from Aldrich Chemical Company, Inc.23.
Purification by preparative HPLC is accomplished as follows. The crude product is taken up in
60 mL of 4 : 1 hexanes : ethyl acetate. The solution is subjected to preparative HPLC
(Note 24) using the same solvent system. Chromatography is monitored by TLC
(Note 26) and the appropriate fractions are combined. Solvent removal with a rotary evaporator at 35°C (12 mm) gives a colorless oil. This material is subjected to bulb-to-bulb distillation at 75–90°C (8 mm).
24.
A Waters Associates Prep LC/System 500 with two cartridges (1.0 kg) of PrepPak-500/Silica was used. The course of the chromatography was followed with a refractive index detector. Approximately 3.6 L of solvent was eluted prior to the product band.
25.
A
Kugelrohr apparatus purchased from the Aldrich Chemical Company, Inc. was used. The receiving bulb was cooled with an ice–water bath. The temperature indicated is the oven temperature.
26.
E. Merck silica gel F-254 plates were used, with
2 : 1 hexanes : ethyl acetate as eluent. Visualization was effected by spraying with a
10% phosphomolybdic acid in ethanol solution followed by heating with a hot air gun.
(2S,3S)-3-Propyloxiranemethanol had an
Rf of ~0.3.
3. Discussion
Both the synthetic
6 and mechanistic
7 aspects of this asymmetric epoxidation process have been reviewed recently. While the process has great scope regarding the allylic alcohol substrate, two classes of substrates present difficulties. These limitations will be best appreciated by reference to the recent reviews;
6,7 however, the main problems are worth mentioning here. When difficulties arise, they are almost never due to the failure of the asymmetric epoxidation process itself but can be traced instead to the nature of the
epoxy alcohol product.
Water-soluble products (e.g., 3- and 4-carbon epoxy alcohols) present obvious isolation problems that have been only partly solved. The other troublesome class of products includes those epoxy alcohols that are unstable under the epoxidation and/or isolation conditions. This latter class consists of the three main types shown below:
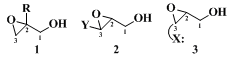
Type
1 is sensitive to nucleophilic opening at the primary epoxide carbon (C-3). Type
2 represents cases where the substituent Y facilitates opening at carbon-3 through resonance stabilization of the incipient carbonium ion. Finally, type
3 includes those cases in which the product bears a heteroatom substituent (X) placed so that a five- or six-membered ring results from anchimerically assisted opening of the epoxide at carbon-3. Not even these structural features (i.e., as in
1, 2, and
3) are always fatal, for some representatives of types
1, 2, and
3 afford good yields of the desired epoxy alcohols.
6,7 Furthermore, when the structure of a given case is marginal, we have found that modification of the epoxidation and/or isolation procedures can lead to substantially improved yields. With such sensitive epoxy alcohols, milder isolation procedures are always employed (see
6 and
8 for a discussion of these modified work up methods). However, certain substrates of types
1, 2, and
3 still fail completely with all current procedures. The best we have been able to do in these difficult cases is to use a strategy that actually takes advantage of the facile epoxide opening process.
6,7,10Questions are often asked concerning the catalytic nature of the asymmetric epoxidation. One notes that the present procedure calls for 50% catalyst. With very favorable substrates, one can realize complete conversion and >95% e.e. using as little as 2% catalyst.
6,7 In the present case, the reaction stops at about 80% conversion using 2% catalyst and almost reaches completion with 10% catalysis.
11 The selection of 50% catalyst is a compromise aimed at rendering the procedure applicable to a wider range of substrates. In the literature, most applications of asymmetric epoxidation use 100% catalyst. This is rarely necessary, but ensures rapid and complete epoxidation in small-scale reactions where cost of the reagents is not an issue. The cases yielding epoxy alcohols that are sensitive to opening require the most catalyst, because the open-diol products are potent inhibitors of the epoxidation catalysis.
6 If one wished to produce molar amounts of a given epoxy alcohol, it would be worthwhile to determine the optimum catalyst loading for the case at hand. In addition to the cost incentive, the isolation procedure becomes simpler as the amount of catalyst is decreased.
The aqueous tartaric acid workup procedure described here is the simplest method for removing the titanium species, but it should be used only with relatively stable epoxy alcohols that are not water-soluble. In this regard, the six-carbon epoxy alcohol made here probably represents the lower limit, as it is on the verge of water solubility. Of course, one cannot assume that this work up will succeed with all six-carbon or larger epoxy alcohols, for in addition to limited water solubility, the product must be fairly resistant to acid-catalyzed epoxide opening processes.
For water-soluble and/or acid-sensitive epoxy alcohols, the "sodium sulfate workup" is generally preferred.
6,8 When making a particularly sensitive epoxy alcohol, one should not only use this
sodium sulfate workup but also modify the initial stage of the epoxidation process so that the reaction mixture only warms to −20°C rather than to 0°C.
Finally, two other practical points are worth mentioning. The early procedures for asymmetric epoxidation called for dilute solutions of
sodium hydroxide to effect tartrate ester hydrolysis. For the reasons given in Note 11, one should always (unless one is certain that the epoxy alcohol is completely insoluble in water) use, instead, NaOH in brine. In this procedure, the excess
tert-butyl hydroperoxide (TBHP) was destroyed early in the workup (FeSO
4), although this is not essential because dilute solutions of TBHP are not dangerous. Other methods for removing excess TBHP in these epoxidations have been reviewed.
6 One of the simplest is to remove it as the azeotrope with
toluene. We have removed up to 0.5 mol of TBHP by this means.
12This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
benzene-d6
hexanes
brine
Eu(hfc)3, tris[3-(heptafluoropropylhydroxymethylene)-d-camphorato] europium(III)
α-methoxy-α-(trifluoromethyl)phenylacetic acid esters
α-methoxy-α-(trifluoromethyl)phenylacetic acid ester
ethanol (64-17-5)
ethyl acetate (141-78-6)
ether (60-29-7)
acetic anhydride (108-24-7)
sodium hydroxide (1310-73-2)
sodium sulfate (7757-82-6)
nitrogen (7727-37-9)
ferrous sulfate (13463-43-9)
pyridine (110-86-1)
toluene (108-88-3)
phosphoric acid (7664-38-2)
tartaric acid (87-69-4)
hexanol (111-27-3)
methylene chloride (75-09-2)
triethylamine (121-44-8)
tert-BUTYL (1605-73-8)
N,N-dimethyl-1,3-propanediamine (109-55-7)
phosphomolybdic acid (51429-74-4)
tert-butyl hydroperoxide (75-91-2)
diethyl tartrate,
(+)-Diethyl L-tartrate
4-dimethylaminopyridine (1122-58-3)
titanium(IV) isopropoxide (546-68-9)
(+)-α-methoxy-α-(trifluoromethyl) phenylacetic acid,
(+)-α-methoxy-α-(trifluoromethyl)phenylacetic acid (56135-03-6)
trans-2-Hexen-1-ol,
E-2-hexen-1-ol (928-95-0)
(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride
epoxy alcohol
m-chloroperoxybenzoic acid (937-14-4)
diethyl (2R,3R)-tartrate (87-91-2)
Oxiranemethanol, 3-Propyl-, (2S,3S)-,
(2S,3S)-3-Propyloxiranemethanol (89321-71-1)
Z-2-hexen-1-ol (928-94-9)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved