Org. Synth. 1989, 67, 86
DOI: 10.15227/orgsyn.067.0086
PALLADIUM-CATALYZED COUPLING OF ACID CHLORIDES WITH ORGANOTIN REAGENTS: ETHYL (E)-4-(4-NITROPHENYL)-4-OXO-2-BUTENOATE
[2-Butenoic acid, 4-(4-nitrophenyl)-4-oxo-, ethyl ester, (E)-]
Submitted by A. F. Renaldo
1, J. W. Labadie, and J. K. Stille
2.
Checked by Robert Aslanian, Cynthia A. Smith, and Andrew S. Kende.
1. Procedure
Caution! Most
tin compounds are toxic,
3;
4;
5 and their preparation should be carried out in a
well-ventilated hood.
A. Tributylethynylstannane. An oven-dried, 2-L, three-necked, round-bottomed flask equipped with a mechanical stirrer, a 100-mL addition funnel, and a nitrogen inlet is charged with 24.0 g (0.26 mol) of lithium acetylide-ethylenediamine complex (Note 1). The system is evacuated, placed under nitrogen, and 800 mL of tetrahydrofuran (Note 2) is added to the system via a cannula. The flask is cooled in an ice-water bath and 70.7 g (0.22 mol) of tributyltin chloride (Note 3) is added dropwise over 45 min. The ice bath is removed and the mixture is stirred for 18 hr at room temperature. The flask is placed in an ice water bath and excess lithium acetylide is hydrolyzed with 20 mL of water. The reaction mixture is concentrated under reduced pressure and washed with hexane (3 × 50 mL). The organic layers are combined and dried over anhydrous magnesium sulfate. Filtration and evaporation of the solvent at reduced pressure gives a colorless oil. Distillation yields 21.4–24.3 g (31–35%) of tributylethynylstannane, bp 90–94°C (0.5 mm) as a water-white liquid (Note 4),(Note 5),(Note 6).
B. (E)-1,2-Bis(tributylstannyl)ethylene. In a 200-mL, one-necked, round-bottomed flask containing a magnetic stirring bar and a nitrogen inlet are placed 20.6 g (0.066 mol) of tributylethynylstannane, 23.1 g (0.079 mol) of tributyltin hydride (Note 7), and 0.25 g (0.0016 mol) of 2,2'-azobis(2-methylpropionitrile) (Note 8). The mixture is heated at 90°C with stirring for 6 hr. Distillation (170–186°C, 0.3 mm) yields 35.1–36.6 g (88–92%) of (E)-1,2-bis(tributylstannyl)ethylene as a clear, colorless oil (Note 9) and (Note 10).
C. Ethyl (E)-3-(tributylstannyl)propenoate. A flame-dried, 1-L, three-necked, round-bottomed flask equipped with a magnetic stirring bar, a 100-mL pressure-equalizing addition funnel (Note 11), and a nitrogen inlet is charged with 26.7 g (0.044 mol) of (E)-1,2-bis(tributylstannyl)ethylene. Tetrahydrofuran (100 mL) is added to the flask by cannula. The system is cooled in a dry ice-acetone bath and the addition funnel is charged, under nitrogen, with 44.8 mL of a 1.2 M solution of methyllithium (0.054 mol) in ethyl ether by means of a double-ended needle (Note 12) and (Note 13). After 10 min the lithium reagent is added dropwise to the flask over a 40-min period. After the addition is complete, the yellow solution is stirred for an additional 2 hr at −78°C during which time a 1-L, one-necked, round-bottomed flask, containing a magnetic stirring bar, is flame-dried under nitrogen. The 1-L, flask, capped with a rubber septum, is charged with a solution of 5.8 g (0.053 mol) of ethyl chloroformate (Note 14) in 150 mL of tetrahydrofuran and cooled to −78°C with a dry ice–acetone bath. Under gentle nitrogen pressure, the metallated reagent is transferred dropwise over a 2.0-hr period by means of a double-ended needle to the 1-L flask containing ethyl chloroformate while the temperature of both flasks is maintained at −78°C (Note 15). After the addition is complete, the reaction mixture is allowed to stir an additional 30 min at −78°C and then treated with 20 mL of methanol in one portion. After 10 min at −78°C the reaction mixture, while still cold, is transferred to a 1-L separatory funnel containing 200 mL of water and 100 mL of hexane. The organic layer is separated and the aqueous layer is washed with hexane (3 × 50 mL). The combined organic layers are dried over anhydrous sodium sulfate, filtered, and concentrated to give a dark-brown oil. The product is dissolved in hexane (30 mL) and purified by chromatography on a column of silica gel (600 g) (Note 16). Elution is carried out initially with hexane (Note 17) and then with hexane/ethyl acetate (95 : 5). Fractions containing the product are combined to give 10.2 g (59%) of ethyl (E)-3-(tributylstannyl)propenoate (Note 18),(Note 19),(Note 20) as a yellow oil.
D. Ethyl (E)-4-(4-nitrophenyl)-4-oxo-2-butenoate. A flame-dried, 150-mL, one-necked, round-bottomed flask containing a magnetic stirring bar and equipped with a side-arm is charged with 3.20 g (17.2 mmol) of p-nitrobenzoyl chloride (Note 21), 0.08 g (0.10 mmol) of benzyl(chloro)bis(triphenylphosphine)palladium(II) (Note 22), and 30 mL of chloroform (Note 23). The bright-yellow solution is evacuated and refilled with carbon monoxide (3 cycles) utilizing a gas bag (Note 24) and (Note 25). After an additional 10 min at room temperature a solution of 8.0 g (20.6 mmol) of ethyl (E)-3-(tributylstannyl)propenoate in 5 mL of chloroform is added to the flask by syringe. The stirring reaction mixture is heated to 50°C for 12 hr while a pressure of 1 atm of carbon monoxide is maintained (Note 26) and (Note 27). The reaction is cooled to room temperature and treated with 18 mL of a 1.2 M solution of pyridinium poly(hydrogen fluoride) (Note 28),(Note 29),(Note 30) along with 10 mL of pyridine. The reaction mixture is allowed to stir at room temperature overnight and then transferred to a 250-mL separatory funnel containing 75 mL of water. After addition of 30 mL of chloroform, the organic layer is washed successively with 10% hydrochloric acid (3 × 20 mL), saturated sodium bicarbonate (3 × 20 mL), water (25 mL), and brine (25 mL). The organic layer is dried over anhydrous sodium sulfate, filtered, and concentrated to give a dark-brown solid. The product is dissolved in chloroform and 15 g of silica gel (Note 16) is added to the solution. Concentration under reduced pressure gives a brown powder of silica coated with product, which is immediately placed on the top of a column of silica gel (50 g) (Note 16). Elution is carried out with ethyl acetate and the fractions are combined and concentrated under reduced pressure (Note 31). The crude product is again placed on a column of silica gel (250 g). Elution is carried out with hexane/ethyl acetate (90 : 10). Fractions containing the product (obtained by collecting the bright yellow band on the column) are combined to give 3.42 g (80%) of ethyl (E)-4-(4-nitrophenyl)-4-oxo-2-butenoate as yellow-green crystals, mp 69–71°C (Note 32).
2. Notes
1.
Lithium acetylide-ethylenediamine complex is purchased from Aldrich Chemical Company, Inc. and used without purification.
2.
Tetrahydrofuran is freshly distilled from
sodium/
benzophenone ketyl at atmospheric pressure under
nitrogen.
3.
Tributyltin chloride, purchased from Alfa Products, Morton/Thiokol, Inc., is distilled immediately before use (bp
128–130°C, 3 mm).
4.
This procedure is a modification of that reported by Seitz.
65.
The remaining fraction of the mixture after distillation was
bis(tributylstannyl)acetylene, which could be recycled for the preparation of
tributylethynylstannane.
6.
The spectral properties are as follows:
1H NMR (270 MHz, CDCl
3) δ: 0.88 (t, 9 H,
J = 7.3), 0.93–1.02 (m, 6 H), 1.25–1.38 (m, 6 H), 1.49–1.60 (m, 6 H), 2.17 (s, 1 H). The infrared spectrum (neat) shows absorption at 3260 and 2000 cm
−1.
7.
Tributyltin hydride is prepared by the procedure of Hayashi
7 in 75% yield on a 0.3-mol scale. The checkers used material from Alfa Products, Morton/Thiokol, Inc., which was vacuum-distilled before use (bp
75–78°C, 0.7 mm).
8.
2,2'-Azobis(2-methylpropionitrile), purchased from Alfa Products, Morton/Thiokol, Inc., is recrystallized from
chloroform prior to use.
9.
The submitters report bp
180–218°C (0.5 mm). The spectral properties are as follows:
1H NMR (270 MHz, CDCl
3) δ: 0.75–1.02 (m, 30 H), 1.21–1.43 (m, 12 H), 1.48–1.63 (m, 12 H), 6.85 (s, 2 H). The infrared spectrum (neat) shows absorption at 1425 and 1020 cm
−1.
10.
(E)-1,2-Bis(tributystannyl)ethylene has been prepared by an alternative procedure using
lithium chloroacetylide.
811.
The funnel is capped with a rubber septum. For ease of operation, volume markings, which correspond to the amount of
methyllithium to be added, are put on the addition funnel.
12.
Caution! Methyllithium is pyrophoric in air; excess quantities of the reagent should be discarded very carefully,13.
Methyllithium is purchased from Aldrich Chemical Company, Inc. Although
butyllithium could also be used in the metallation step, a cleaner product is obtained with
methyllithium.
14.
Ethyl chloroformate, purchased from Aldrich Chemical Company, Inc., is distilled at atmospheric pressure prior to use, discarding the first 25 mL.
15.
The solution in the flask which contains the
ethyl chloroformate is bright yellow and gradually becomes dark red on the addition of the metallated reagent.
16.
The checkers used
Kieselgel 60 (230–400 mesh), purchased from E. Merck. The submitters used
silica gel (32–63 mesh) purchased from Universal Scientific, Inc.
17.
Hexane removes the
methyltributyltin.
18.
The spectral properties are as follows:
1H NMR (270 MHz, CDCl
3) δ: 0.78–0.99 (m, 12 H), 1.01–1.49 (m, 18 H), 4.11 (q, 2 H
J = 7.3), 6.22 (d, 1 H,
J = 19.7), 7.65 (d, 1 H,
J = 19.6). The infrared spectrum (neat) shows absorption at 1715, 1580, and 1200 cm
−1.
19.
Attempts to purify the product by vacuum distillation, bp
110–138°C (0.05 mm), result in
7–8% isomerization to
ethyl (Z)-3-(tributylstannyl)propenoate [based on
1H NMR (270 MHz) analysis].
20.
The product should be stored under
nitrogen at 0°C to prevent decomposition.
21.
p-Nitrobenzoyl chloride, purchased from Aldrich Chemical Company, Inc., is recrystallized from
hexane prior to use.
22.
Benzyl(chloro)bis(triphenylphosphine)palladium(II) is prepared from tetrakis(triphenylphosphine)palladium(0)9 (also available from Aldrich Chemical Company, Inc.) by the procedure of Fitton.
1023.
Chloroform is freshly distilled at atmospheric pressure under
nitrogen and filtered through a plug of neutral alumina.
24.
The bright-yellow color of the solution changes to light green after saturation with
carbon monoxide. The presence of
carbon monoxide prevents decarbonylation of the acylpalladium complex and thus the formation of
ethyl p-nitrocinnamate.
25.
The gas bag is purchased from Fisher Scientific.
26.
The pressure of 1 atm is maintained by use of the gas bag.
27.
The reaction changes color from light green to bright orange.
28.
Pyridinium poly(hydrogen fluoride) is purchased from Aldrich Chemical Company, Inc.29.
The solution of
pyridinium poly(hydrogen fluoride) in
tetrahydrofuran and
pyridine is prepared according to the procedure of Trost.
11 Pyridine is freshly distilled over
calcium hydride at atmospheric pressure and stored over Linde 4A molecular sieves.
30.
The orange reaction mixture changes to deep red and the reaction becomes slightly exothermic (50–60°C).
31.
The initial filtration with silica gel is necessary to remove most of the
tributyltin fluoride.
32.
The spectral properties are as follows:
1H NMR (270 MHz, CDCl
3) δ: 1.34 (t, 3 H,
J = 7.2), 4.30 (q, 2 H,
J = 7.2), 6.92 (d, 1 H,
J = 15.6), 7.85 (d, 1 H,
J = 15.6), 8.13 (d, 2 H,
J = 8.9), 8.35 (d, 2 H,
J = 8.8);
13C NMR (68 MHz, CDCl
3) δ: 14.2, 61.7, 124.1, 129.9, 134.3, 135.5, 141.4, 151.0, 165.1, 188.4. The infrared spectrum (Nujol) shows the following absorption cm
−1: 1690, 1660, 1590, 1300, 990, 970, 710.
3. Discussion
The procedure for synthesis of the title compound is representative of the palladiumcatalyzed coupling of acid chlorides with organotin reagents.
12 13 14 15 The formation of ketones by Grignard reagents,
16 organocuprates,
17 18 and organoborates
19 has been reported. The principal advantage of the palladium-catalyzed organotin coupling method lies in the broad range of functionality that can be introduced in the product. The reaction can be carried out under mild, neutral conditions with functional groups on the acid chloride such as nitro, nitrile, haloaryl, methoxy, ester, and even aldehyde.
15 Solvents other than
chloroform, such as
tetrahydrofuran,
hexamethylphosphorictriamide, and
dichloromethane, can be used in this reaction.
The unsymmetric tetraorganotin reagent has been demonstrated to transfer selectively the vinyl group rapidly without butyl transfer occurring. By using a tributyl or trimethyl organotin reagent (e.g., Bu
3SnR or Me
3SnR), the order of transfer of the R groups is RC≡C- > RCH=CH- > Ar- > RCH=CH-CH
2- > ArCH
2- > CH
3OCH
2- > C
nH
2n + 1.
20 21A number of functionalized organotin derivatives have been used in palladium-catalyzed coupling to produce aromatic heterocyclic ketones,
22 acetylenic ketones,
23 and vinyl ketones.
24 25 26 The organotin coupling method has been used effectively in the preparation of a key methyl ketone intermediate in the total synthesis of
(±)-quadrone27 and in the preparation of
5, a key precursor in the synthesis of the antibiotic
pyrenophorin 6 (Eq. 1).
21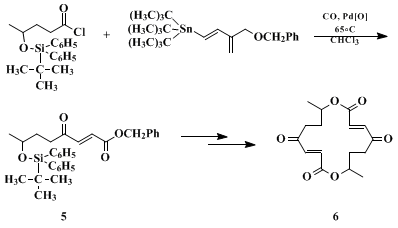
The organotin reagents are very stable since they can withstand distillation as well as chromatography on silica gel. The procedure for preparation of
tributylethynylstannane (1) in Part A is based on one reported by Bottaro et al.
6 Bis(tributylstannyl)ethylene (2) has been prepared from
lithium chloroacetylide8 and
tributylethynylstannane.
28 29 Although
ethyl (E)-3-(tributylstannyl)propenoate (3) is produced from transmetallation
30 of
2 or hydrostannation
31 of
ethyl propiolate, other known procedures to synthesize
3 include conjugate addition to
tributylstannylcuprate32 33 to
ethyl propiolate, and
tributylstannylcopper to β-substituted acrylates.
34In most cases the trimethyltin reagents are preferred since the by-product,
trimethyltin chloride, can easily be removed by water extraction. In the case of the water-insoluble
tributyltin chloride it is necessary to add an aqueous solution of
potassium fluoride to an ethereal solution of the product, thereby forming insoluble
tributyltin fluoride, which can be separated by filtration.
20,21,35 However, a completely homogeneous and neutral fluoride source,
pyridinium hydrofluoride,
11 is used in this procedure, making the filtration unnecessary and simplifying the subsequent chromatography step.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
benzophenone ketyl
pyrenophorin
lithium acetylide-ethylenediamine
pyridinium poly(hydrogen fluoride
(E)-1,2-Bis(tributystannyl)ethylene
pyridinium poly(hydrogen fluoride)
(±)-quadrone
hydrochloric acid (7647-01-0)
ethyl acetate (141-78-6)
methanol (67-56-1)
ethyl ether (60-29-7)
carbon monoxide (630-08-0)
chloroform (67-66-3)
sodium bicarbonate (144-55-8)
sodium sulfate (7757-82-6)
nitrogen (7727-37-9)
tin (7440-31-5)
pyridine (110-86-1)
sodium (13966-32-0)
dichloromethane (75-09-2)
ethyl chloroformate (541-41-3)
lithium (7439-93-2)
magnesium sulfate (7487-88-9)
butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
potassium fluoride (7789-23-3)
hexane (110-54-3)
Methyllithium (917-54-4)
tributyltin hydride (688-73-3)
calcium hydride (7789-78-8)
ethyl propiolate (623-47-2)
hexamethylphosphorictriamide (680-31-9)
tetrakis(triphenylphosphine)palladium(0) (14221-01-3)
trimethyltin chloride (1066-45-1)
Lithium acetylide,
Lithium acetylide-ethylenediamine complex (6867-30-7)
tributyltin chloride (1461-22-9)
Tributylethynylstannane (994-89-8)
benzyl(chloro)bis(triphenylphosphine)palladium(II)
bis(tributylstannyl)acetylene (994-71-8)
lithium chloroacetylide
methyltributyltin (1528-01-4)
tributyltin fluoride
Bis(tributylstannyl)ethylene
tributylstannylcuprate
tributylstannylcopper
pyridinium hydrofluoride
p-Nitrobenzoyl chloride (122-04-3)
Ethyl (E)-4-(4-nitrophenyl)-4-oxo-2-butenoate,
2-Butenoic acid, 4-(4-nitrophenyl)-4-oxo-, ethyl ester, (E)- (131504-53-5)
(E)-1,2-Bis(tributylstannyl)ethylene (14275-61-7)
Ethyl (E)-3-(tributylstannyl)propenoate (106335-84-6)
ethyl (Z)-3-(tributylstannyl)propenoate
ethyl p-nitrocinnamate
2,2'-azobis(2-methylpropionitrile)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved