Org. Synth. 2000, 77, 78
DOI: 10.15227/orgsyn.077.0078
DIASTEREOSELECTIVE SYNTHESIS OF PROTECTED VICINAL AMINO ALCOHOLS: (S)-2-[(4S)-N-tert-BUTOXYCARBONYL-2,2-DIMETHYL-1,3-OXAZOLIDINYL]-2-tert-BUTYLDIMETHYLSILOXYETHANAL FROM A SERINE-DERIVED ALDEHYDE
[
3-Oxazolidinecarboxylic acid, 4-[1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-oxoethyl]-2,2-dimethyl-, 1,1-dimethylethyl ester, [S-(R*,R*)]-
]
Submitted by Alessandro Dondoni and Daniela Perrone
1
.
Checked by Hou Chen and William R. Roush.
1. Procedure
A.
(S)-2-{[(4S)-N-tert-Butoxycarbonyl-2,2-dimethyl-1,3-oxazolidin-4-yl]hydroxymethyl}-1,3-thiazole
(2)
. A
100-mL, two-necked, round-bottomed flask, containing a magnetic stirring bar, is equipped with a low-temperature thermometer and one equalizing dropping funnel
(Note 1). The funnel is charged with a solution of
2-(trimethylsilyl)thiazole (2-TST, 6.17 g, 39.2 mmol, (Note 2)) in
10 mL of methylene chloride
(Note 3) and connected to a
nitrogen flow line. The flask is charged with a solution of aldehyde
1
(7.5 g, 32.7 mmol, (Note 4)) in
65 mL of methylene chloride
, then cooled to −35°C in a
CryoCool bath and 2-TST is added dropwise over 15 min
(Note 5). After completion of the addition, the funnel is washed with two
1-mL portions of methylene chloride
, then replaced with a rubber septum. The flask is allowed to stand at −20°C (freezer) overnight (15 hr,
(Note 6)), then allowed to warm to room temperature and concentrated under reduced pressure. To the residue dissolved in
40 mL of tetrahydrofuran
is added, in one portion,
tetrabutylammonium fluoride trihydrate (12.3 g, 39.0 mmol), and the brown reaction mixture is stirred at room temperature for 30 min
(Note 7) and
(Note 8). The solvent is removed under reduced pressure and the residue is partitioned between
ethyl acetate (200 mL) and
aqueous saturated sodium bicarbonate solution (2 × 250 mL). The aqueous phase is extracted with
ethyl acetate (3 × 80 mL); then the combined organic phases are dried with
anhydrous sodium sulfate
and concentrated under reduced pressure using a
1-L flask to give
9.76 g (
95% crude yield) of a clear brown solid. Analysis of crude product by
1H NMR indicates a chemical purity of 90-95% and a diastereomeric ratio of 92% (anti-adduct). To the same 1-L flask equipped with a
reflux condenser is added
475 mL of cyclohexane
. The mixture is warmed at 80°C (water bath) until an homogeneous solution is obtained
(Note 9). The hot solution is quickly filtered and allowed to cool to room temperature and to stand at this temperature overnight, at which time a white precipitate is formed. The precipitate is collected by filtration and washed with three
10-mL portions of cold cyclohexane
to give
7.15-7.50 g (
70-73% yield) of
2
as a white solid: mp
171-173°C
(Note 10). The filtrate is concentrated under reduced pressure and purified by flash chromatography with a
5 × 14-cm column of silica gel
(Note 11),
(cyclohexane-ethyl acetate (2:3) and 0.2% of triethylamine), to give an additional
1.20-1.40 g (
11.6-13.6% yield) of a mixture of syn- and anti-
(2)
adducts
(Note 12). Analysis of this mixture by
1H NMR indicates a syn/anti diastereomeric ratio of 1.2:1.
B.
(S)-2-{[(4S)-N-tert-Butoxycarbonyl-2,2-dimethyl-1,3-oxazolidin-4-yl]-tert-butyldimethylsiloxy}-1,3-thiazole
(3)
. A
100-mL, one-necked, round-bottomed flask
(Note 1) equipped with a
magnetic stirring bar and a rubber septum is charged with a solution of the alcohol
2
(4.0 g, 12.7 mmol),
99%
triethylamine (3.54 mL, 25.5 mmol), and
4-dimethylaminopyridine (DMAP, 0.12 g, 0.98 mmol) in
30 mL of N,N-dimethylformamide
(Note 13) and
(Note 14). While the solution is stirred at room temperature,
tert-butyldimethylsilyl trifluoromethanesulfonate (4.38 mL, 19.1 mmol, (Note 15)) is introduced into the reaction flask through the rubber septum using a syringe over a period of 1 min.
Caution! tert-Butyldimethylsilyl trifluoromethanesulfonate is a toxic compound that should be handled in a well-ventilated fume hood. After stirring at room temperature for 1 hr
(Note 16),
4 mL of methanol
is added via syringe. The reaction solution is stirred for an additional 30 min, then the solvent is removed under reduced pressure. The residue is dissolved in
60 mL of hexanes-ethyl acetate (4:1) and washed with
100 mL of aqueous saturated sodium chloride (NaCl). The organic phase is dried over
anhydrous sodium sulfate
and concentrated under reduced pressure to give
5.18-5.30 g (
95-97% crude yield) of
3
as a colorless syrup
(Note 17). Analysis of crude product by
1H NMR indicates a chemical purity of > 98%.
C.
(S)-2-[(4S)-N-tert-Butoxycarbonyl-2,2-dimethyl-1,3-oxazolidin-4-yl]-2-tert-butyldimethylsiloxyethanal
(4)
. A
100-mL, one-necked, round-bottomed flask, equipped with a 2-cm egg-shaped magnetic stirring bar
(Note 18), is charged with a mixture of the
O-silyl ether
3
(2.0 g, 4.66 mmol), activated 4 Å powdered molecular sieves (9.33 g), and
47 mL of acetonitrile
(Note 19) and
(Note 20). The suspension is stirred at room temperature for 10 min, then
methyl triflate (0.63 mL, 5.6 mmol, (Note 21)) is added via syringe.
Caution! Methyl triflate is toxic and a suspected carcinogen that should be handled in a well-ventilated fume hood. The mixture is vigorously stirred for 15 min and concentrated to dryness under reduced pressure
(Note 22). To the residue suspended in
47 mL of 1:1 methanol/diethyl ether
and cooled with an
ice-water bath is added under vigorous stirring
sodium borohydride (0.39 g, 10.3 mmol, (Note 23)). Upon completion of the addition, the flask is removed from the ice bath and the solution is stirred at room temperature for 15 min, diluted with
acetone (4 mL) and filtered through a
Celite pad. The pad is rinsed with three
47-mL portions of acetone
and the filtrate is concentrated under reduced pressure
(Note 24). The residue is dissolved in
47 mL of acetonitrile-water (10:1) and the flask is placed in an
ultrasonic cleaning bath and treated with
98%
copper(II) oxide (2.97 g, 37.3 mmol, (Note 25)) and then with
95%
copper(II) chloride dihydrate (0.79 g, 4.63 mmol, (Note 25)). After 10 min, the mixture is filtered through a Celite pad that is rinsed with four
47-mL portions of acetonitrile
. The filtrate is concentrated under reduced pressure
(Note 24) to give a brown syrup. The brown residue is sonicated for 5 min each with five
47-mL portions of diethyl ether
, in an ultrasonic cleaning bath. The liquid layer is pipetted and filtered through a
1 × 6.5-cm (h × d) Florisil (100-200 mesh) pad. The filtrate appears almost colorless and is concentrated under reduced pressure
(Note 24), to give
1.4-1.52 g (
80-87% crude yield) of crude aldehyde
4
as a clear yellow oil
(Note 26). Analysis of the crude product by
1H NMR indicates a chemical purity of 90-95%. Crude
4
can be purified by flash chromatography over a
4.5 × 15-cm column of Silica Gel 60
(Note 11) eluting with
cyclohexane-ethyl acetate (9:1) to give
1.3-1.4 g (
75-80% yield) of pure aldehyde
(Note 27).
2. Notes
1.
The glass components of the apparatus were dried overnight in a 150°C oven and allowed to cool in a
desiccator over a drying agent before assembly.
2.
2-(Trimethylsilyl)thiazole (2-TST) is prepared according to the Organic Syntheses procedure;2 it is also commercially available (Acros Chimica or Aldrich Chemical Company, Inc.). The commercial product was purified by distillation prior to use.
3.
Methylene chloride was dried before use by distillation from
calcium hydride
under an atmosphere of
nitrogen.
4.
The preparation of the
N-Boc-L-serinal acetonide
is described in the accompanying procedure,
p. 64. The crude aldehyde was used without further purification.
5.
During the addition the clear yellow solution became red-orange.
6.
After standing for 15 hr at −20°C the reaction solution returned to clear yellow.
7.
Tetrahydrofuran was dried before use by distillation from
sodium metal and benzophenone
under an atmosphere of
nitrogen.
8.
Tetrabutylammonium fluoride trihydrate was purchased from Acros Chimica
and used without further purification.
9.
The checkers found it necessary to heat the solution to reflux to dissolve the solid. The hot solution was filtered by pouring into a preheated glass funnel containing a plug of glass wool to remove insoluble materials.
10.
The product exhibits the following properties:
[α]
D
−51.2° (CHCl
3,
c 1.7), IR (KBr) cm
−1: 3230, 1695, 1656
;
1H NMR (300 MHz, DMSO-d
6, 120°C) δ: 1.38 (s, 9 H), 1.46 (s, 3 H), 1.56 (s, 3 H), 3.82 (dd, 1 H, J = 9.0, 6.8), 3.95 (dd, 1 H, J = 9.0, 3.0), 4.26 (ddd, 1 H, J = 6.4, 4.1, 3.0), 5.15 (dd, 1 H, J = 5.5, 4.1), 6.11 (d, 1 H, J = 5.5, ex D
2O), 7.55 (d, 1 H, J = 3.0), 7.71 (d, 1 H, J = 3.0)
;
13C NMR (75 MHz, CDCl
3) δ: 24.5, 25.7, 28.2, 62.2, 64.4, 73.4, 81.5, 94.6, 119.0, 142.4, 154.5, 172.6
; HRMS, calcd for C
14H
23N
2O
4S [M+H]
+ 315.1378, found 315.1379
Purification with flash chromatography on
silica gel eluting with (1:1)
cyclohexane-
ethyl acetate gave a product with a maximum rotation of
[α]
D
−54.1° (CHCl
3,
c 1.0) and a mp of
174-175°C.
11.
Silica Gel 60 (230-400 mesh) was obtained from Merck & Company, Inc.
12.
Recrystallization of this mixture from
cyclohexane (70 mL) gave ca.
0.40 g of
2
(
4%).
13.
Triethylamine (99%) and 4-dimethylaminopyridine were purchased from Acros Chimica
and used without further purification.
14.
N,N-Dimethylformamide was purchased from Acros Chimica
and dried over activated 4 Å molecular sieves (8-12 mesh, Acros) before use.
15.
tert-Butyldimethylsilyl trifluoromethanesulfonate was purchased from Acros Chimica
and used as received under an atmosphere of
nitrogen.
16.
TLC analysis on
Silica Gel 60F-254 plates eluting with
cyclohexane-
ethyl acetate (7:3) showed the clean formation of product with R
f = 0.69, at the expense of the starting material with R
f = 0.3. If a small amount of starting material was still present at this time, more
tert-butyldimethylsilyl trifluoromethanesulfonate (0.22 mL, 0.95 mmol) and
0.2 mL of triethylamine
were added and the reaction mixture was stirred for a further 30 min, at which time the TLC analysis generally showed the reaction to be complete.
17.
The crude product exhibits the following properties:
[α]
D
−46° (CHCl
3,
c 0.65); IR (KBr) cm
−1
1705, 1685
;
1H NMR (300 MHz, DMSO-d
6, 120°C) δ: −0.04 (s, 3 H), 0.10 (s, 3 H), 0.94 (s, 9 H), 1.41 (s, 9 H), 1.45 (s, 3 H), 1.60 (s, 3 H), 3.80 (dd, 1 H, J = 8.9, 7.0), 4.09 (dd, 1 H, J = 8.9, 3.5), 4.17 (dt, 1 H, J = 7.0, 3.5), 5.55 (d, 1 H, J = 3.5), 7.60 (d, 1 H, J = 3.1), 7.75 (d, 1 H, J = 3.1)
; HRMS, calcd for C
20H
37N
2O
4SSi [M+H]
+ 429.2243, found 429.2229
. Purification with flash chromatography on
silica gel eluting with
cyclohexane-
ethyl acetate (9:1) gave a product with a maximum rotation of
[α]
D
−47.6° (CHCl
3,
c 0.60). Upon cold storage a chromatographed sample of
3
crystallized: mp
53-55°C.
18.
For efficient stirring a powerful magnetic stirrer should be used.
19.
Activated 4 Å molecular sieves (powder, < 5 micron) were purchased from Aldrich Chemical Company, Inc., and used as received.
20.
Acetonitrile was purchased from Acros Chimica
and dried over activated 4 Å molecular sieves (8-12 mesh, Acros) before use.
21.
Methyl triflate was purchased from Aldrich Chemical Company, Inc.
, and used as received.
22.
TLC analysis on Silica Gel 60F-254 plates eluting with
cyclohexane-
ethyl acetate (9:1) showed the formation of product with R
f = 0, at the expense of the starting material.
23.
Sodium borohydride was purchased from Aldrich Chemical Company, Inc.
, and used without further purification.
24.
Bath temperature should not exceed 40°C.
25.
Copper(II) oxide and copper(II) chloride dihydrate were purchased from Acros Chimica
and used without further purification. The checkers found that the yield of Step C was substantially reduced (
64-65%) when an old bottle of CuO was used. Results identical to those reported by the submitters were obtained using a new bottle of this reagent.
26.
The crude product exhibits the following properties:
[α]
D
−40.7° (CHCl
3,
c, 0.6); IR (neat) cm
−1: 1740, 1712, 1690
;
1H NMR (300 MHz, DMSO-d
6, 120°C) δ: 0.1 (s, 6 H), 0.95 (s, 9 H), 1.44 (s, 9 H), 1.49 (s, 3 H), 1.54 (s, 3 H), 3.88 (dd, 1 H, J = 9.0, 2.5), 3.95 (dd, 1 H, J = 9.0, 5.9), 4.07 (dt, 1 H, J = 6.0, 2.5), 4.26 (dd, 1 H, J = 6.0, 2.0), 9.55 (d, 1 H, J = 2.0)
; HRMS, calcd for C
18H
36NO
5Si [M+H]
+ 374.2362, found 374.2357
.
27.
After flash chromatography the product showed a maximum rotation of
[α]
D
−45.7° (CHCl
3,
c 0.7).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The 2-amino-1,3-diol moiety is a structural unit featured by numerous natural products and synthetic analogues. For instance it is present at the polar head of sphingosines,
3 and at the non-reducing end of aza sugars.
4 Therefore chiral 3-amino-2,4-dihydroxybutanals are potential building blocks for the synthesis of those biologically active compounds.
5
6
7 Since either R or S configuration at the stereocenters may be required, it is advisable to rely on a synthetic method that allows one to obtain these building blocks with all possible absolute and relative stereochemical arrangements. Accordingly, the submitters have approached the synthesis of these compounds by two routes (Scheme 1), which are both centered on the use of the thiazole ring as a convenient precursor of the formyl group (Thiazole Aldehyde Synthesis).
8
9
10 These routes are complementary in the sense that starting from the same
L-serine derived oxazolidine methyl ester
I they lead to either R or S diastereomer oxazolidine alcohol
VI. The S-configuration of the
amino ester is employed for the construction of the new stereocenter through internal asymmetric induction. Thus, in one route ester
I is reduced to the oxazolidine aldehyde
II, which then undergoes stereoselective anti-addition of the thiazole-bearing organosilane
III to give the S-oxazolidine alcohol
VIa as major product (aldehyde route).
5,11 In the other route the same ester
I is first converted into the oxazolidine ketone
V through nucleophilic substitution with another metalated thiazole
IV. Ketone
V is then reduced stereoselectively to give the R-oxazolidine alcohol
VIb as the major product (ketone route).
7,12 The opposite stereochemical outcome observed in this case is determined by the stereoselective hydride anti-addition to the carbonyl. The thiazole bearing diastereomeric oxazolidine alcohols
VIa and
VIb, after suitable protection of the hydroxy group, are converted into the corresponding aldehydes
VIIa and
VIIb by the usual thiazole-to-formyl cleavage protocol.
13 Evidently the antipodes of these aldehydes can also be prepared by the same routes starting from the
D-serine derived enantiomer of ester
I. Both routes can be scaled-up for the synthesis of gram quantities of these aldehydes. The thiazole ring serves as a convenient auxiliary in this methodology as well, since it tolerates different types of reaction conditions without any interference, and when needed it is readily converted into the formyl group. The thiazole-to-formyl cleavage occurs under mild and neutral conditions that do not affect the stereochemical integrity and chemical stability of the resulting aldehyde. Crude compound
VII obtained by either the aldehyde or the ketone route is pure enough for use as a synthetic chiral building block.
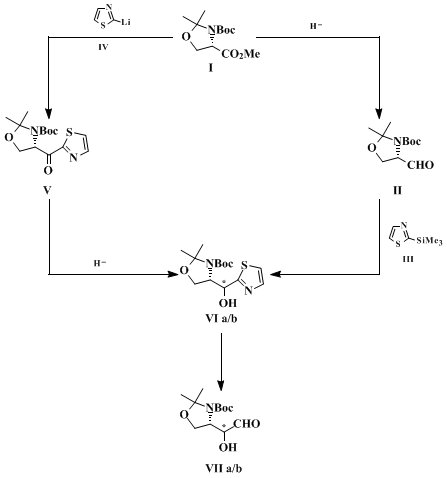
This procedure describes an example of the "aldehyde route". The addition of
2-(trimethylsilyl)thiazole (2-TST) to aldehydes occurs readily and does not require the presence of a fluoride ion source.
14 The resulting secondary alcohol is as a rule isolated in very good yield. The sense of the diastereofacial selectivity of the addition reaction to chiral α-amino aldehydes can be controlled by differential protection of the amino group. Specifically, compounds with double protection afford anti-amino alcohols, whereas those with single protection give syn-diastereomers.
5,11 Accordingly the addition of 2-TST to aldehyde
1
affords alcohol
2
as almost a single product (Step A). Rapid purification of this compound from the syn-isomer (≤ 8%) is carried out by crystallization from
cyclohexane. Pure alcohol
2
is then converted almost quantitatively into the tert-butyldimethylsilyl ether
3
(Step B). Other types of protection of the hydroxy group of
3
can also be applied. Finally crude O-silyl ether
3
is subjected to the conventional thiazole-to-formyl deblocking protocol
13 (Step C) that is carried out through a sequence of very effective reactions, i.e., N-methylation, reduction, and metal-catalyzed hydrolysis. This aldehyde liberation requires no more than 4 hr of work. Isolated crude compound
4
is obtained in
52-59% yield from aldehyde
1.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2-(Trimethylsilyl)thiazole:
Thiazole, 2-(trimethylsilyl)- (10); (79265-30-8)
(S)-2-{[(4S)-N-tert-Butoxycarbonyl-2,2-dimethyl-1,3-oxazolidin-4-yl]hydroxymethyl}-1,3-thiazole:
3-Oxazolidinecarboxylic acid, 4-(hydroxy-2-thiazolylmethyl)-2,2-dimethyl-, 1,1-dimethylethyl ester, [(S-(R,R)]- (12); (115822-48-5)
1,1-Dimethylethyl (S)-4-formyl-2,2-dimethyloxazolidinecarboxylate:
3-Oxazolidinecarboxylic acid, 4-formyl-2,2-dimethyl-, 1,1-dimethylethyl ester, (S)- (11); (102308-32-7)
Tetrabutylammonium fluoride trihydrate:
Ammonium, tetrabutyl-, fluoride, hydrate (8,9); (22206-57-1)
(S)-2-{[(4S)-N-tert-Butoxycarbonyl-2,2-dimethyl-1,3-oxazolidin-4-yl]-tert-butyldimethylsiloxy}-1,3-thiazole:
3-Oxazolidinecarboxylic acid, 4-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-thiazolylmethyl]2,2-dimethyl-, 1,1-dimethylethyl ester, [S-(R,R)]- (13) (168326-00-9)
Triethylamine (8);
Ethanamine, N,N-diethyl- (9); (121-44-8)
4-Dimethylaminopyridine: HIGHLY TOXIC:
Pyridine, 4-(dimethylamino)- (8);
4-Pyridinamine, N,N-dimethyl- (9); (1122-58-3)
N,N-Dimethylformamide: CANCER SUSPECT AGENT:
Formamide, N,N-dimethyl- (8,9); (68-12-2)
tert-Butyldimethylsilyl trifluoromethanesulfonate:
Methanesulfonic acid, trifluoro-, (1,1-dimethylethyl)dimethylsilyl ester (10); (69739-34-0)
(S)-2-2-[(4S)-N-tert-Butoxycarbonyl-2,2-dimethyl-1,3-oxazolidin-4-yl-2-tert-butyldimethylsiloxyethanal:
3-Oxazolidinecarboxylic acid, 4-[1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-oxoethyl]-2,2-dimethyl-, 1,1-dimethylethyl ester, [S-(R*,R*)]- (13); (192119-88-3)
Acetonitrile (8,9); (75-05-8)
Methyl triflate:
Methyl trifluoromethanesulfonate:
Methanesulfonic acid, trifluoro-, methyl ester (8,9); (333-27-7)
Sodium borohydride:
Borate (1−), tetrahydro-, sodium (8,9); (16940-66-2)
Copper(II) oxide:
Copper oxide (8,9); (1317-38-0)
Copper(II) chloride dihydrate:
Copper chloride dihydrate (8);
Copper chloride, dihydrate (9); (10125-13-0)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved