Org. Synth. 1971, 51, 11
DOI: 10.15227/orgsyn.051.0011
ALDEHYDES FROM ACID CHLORIDES BY REDUCTION OF ESTER-MESYLATES WITH SODIUM BOROHYDRIDE: CYCLOBUTANECARBOXALDEHYDE
Submitted by M. Ross Johnson and Bruce Rickborn
1.
Checked by Saul C. Cherkofsky and Richard E. Benson.
1. Procedure
Caution! Reactions and subsequent operations involving peracids and peroxy compounds should be run behind a safety shield. For relatively fast reactions, the rate of addition of the peroxy compound should be slow enough so that it reacts rapidly and no significant unreacted excess is allowed to build up. The reaction mixture should be stirred efficiently while the peroxy compound is being added, and cooling should generally be provided since many reactions of peroxy compounds are exothermic. New or unfamiliar reactions, particularly those run at elevated temperatures, should be run first on a small scale. Reaction products should never be recovered from the final reaction mixture by distillation until all residual active oxygen compounds (including unreacted peroxy compounds) have been destroyed. Decomposition of active oxygen compounds may be accomplished by the procedure described in Korach, M.; Nielsen, D. R.; Rideout, W. H. Org. Synth. 1962, 42, 50 (Org. Synth. 1973, Coll. Vol. 5, 414). [Note added January 2011].
A. erythro-2,3-Butanediol monomesylate. A 2-l., round-bottomed flask is equipped with a 1-l. dropping funnel attached to a calcium chloride drying tube. A magnetic stirring bar is placed in the flask and a solution of 48.0 g. (0.500 mole) of methanesulfonic acid (Note 1) in 500 ml. of anhydrous diethyl ether is added. Stirring is begun, and the flask is cooled in an ice-water bath while a solution of 37 g. (0.52 mole) of trans-2-butene oxide (Note 2) and (Note 3) in 500 ml. of anhydrous ether is added over a period of 3–4 hours (Note 4). After 6 hours the cooling bath is removed and the mixture is stirred an additional 12 hours. The ether and any excess epoxide are removed with a rotary evaporator at 25° and water aspirator pressure, giving 83–84 g. (99–100%) of erythro-2,3-butanediol monomesylate as a clear, colorless, somewhat viscous liquid (Note 5).
B. erythro-3-Methanesulfonyloxy-2-butyl cyclobutanecarboxylate. A 500-ml., round-bottomed flask, cooled in an ice-water bath, equipped with a 50-ml. dropping funnel and a magnetic stirring bar is charged with 35.3 g. (0.210 mole) of erytho-2,3-butanediol monomesylate and 150 ml. of dry pyridine. Stirring is begun, and 23.7 g. (0.200 mole) of cyclobutanecarboxylic acid chloride (Note 6) is added over a period of 1 hour. The cooling bath is removed, and stirring is continued for 8 hours. The mixture is added to 500 ml. of ether, and the resulting solution washed with three 250-ml. portions of 3 N sulfuric acid. The pyridine-free solution is washed with 250 ml. of a saturated sodium hydrogen carbonate solution and then with 250 ml. of water. The ether solution is dried over 2 g. of anhydrous magnesium sulfate. The solvent is removed with a rotary evaporator at 25°, giving 45.1–48.0 g. (90–96%) of erythro-3-methanesulfonyloxy-2-butyl cyclobutanecarboxylate as a pale yellow, viscous liquid (Note 7).
C. 2-Cyclobutyl-cis-4-trans-5-dimethyl-1,3-dioxolane. A 2-l., three-necked, round-bottomed flask is equipped with a mechanical stirrer, a 125-ml. dropping funnel, and a condenser attached to a nitrogen line with a bubbler device to permit maintenance of a positive pressure of nitrogen. Anhydrous pyridine (650 ml.) and 5 g. (0.1 mole) of sodium borohydride (Note 8) are added to the flask, stirring is begun, and the mixture is heated at reflux. A solution of 25 g. (0.10 mole) of erythro-3-methanesulfonyloxy-2-butyl cyclobutanecarboxylate in 50 ml. of anhydrous pyridine is added from the dropping funnel over a period of 30 minutes, and heating at reflux is continued for 8 hours. After cooling, 50 ml. of water is added (some heat is evolved), and the mixture is transferred to a 4-l. separatory funnel with 1 l. of pentane (Note 9), and 700 ml. of cold 3 N sulfuric acid saturated with sodium chloride. The aqueous layer is separated and washed with two 250-ml. portions of pentane. The pentane extractions are combined and washed with three 500-ml. portions of 3 N sulfuric acid saturated with sodium chloride and finally with 500 ml. of saturated sodium hydrogen carbonate. The pentane solution is dried over 1 g. of anhydrous potassium carbonate and evaporated on a steam bath. The product is distilled through a short Vigreux column, yielding 6.7–7.6 g. (43–49% (Note 10)) of 2-cyclobutyl-cis-4-trans-5-dimethyl-1,3-dioxolane, b.p. 79–83° (22 mm.) (Note 11).
D. Cyclobutanecarboxaldehyde. A 1-l., round-bottomed flask equipped with a magnetic stirring bar and a 60-cm. glass helix-packed column is charged with 600 ml. of 3 N sulfuric acid, 200 ml. of N,N-dimethylformamide (Note 12), and 20 g. (0.13 mole) of 2-cyclobutyl-cis-4-trans-5-dimethyl-1,3-dioxolane. The mixture is heated to gentle reflux, and cyclobutanecarboxaldehyde is collected as a steam distillate, b.p. 86°. After the distillation of the oil has ceased, the product is transferred to a separatory funnel, and the lower layer of water is discarded. The oil is dissolved in 25 ml. of ether and dried over anhydrous sodium sulfate. The product is distilled through a small Vigreux column. After removal of the ether, 6.2–6.7 g. (58–63%) of cyclobutanecarboxaldehyde is collected, b.p. 56–59° (120 mm.) (Note 13).
2. Notes
1.
Methanesulfonic acid was obtained from Aldrich Chemical Company, Inc., and distilled prior to use. The fraction collected at 140° (0.2 mm.) was used.
2.
trans-2-Butene oxide was prepared by appropriate modification of the procedure in
Org. Synth., Coll. Vol. 4, 860 (1963). A
2-l., four-necked, round-bottomed flask fitted with a mechanical stirrer, a 1-l. dropping funnel, an
acetone–dry ice condenser, and a
thermometer is charged with
1 l. of 1,1,2,2-tetrachloroethane. The condenser is packed with dry ice and
acetone, and the flask is cooled in a
methanol-ice bath to −15°.
trans-2-Butene (153 g., 2.73 moles) (Phillips Petroleum Company, 99%) is distilled into the flask from a tared, chilled
trap. Six hundred milliliters of
40% peracetic acid (FMC Corporation), to which has been added
30 g. sodium acetate to neutralize the
sulfuric acid present, is added to the stirred solution from the dropping funnel over a period of 2 hours. The mixture is stirred at −15° for another hour, then allowed to warm to room temperature. The mixture is poured into 1 l. of ice-cold water. The organic layer is separated, washed first with
10% sodium carbonate solution, then with water, dried over
magnesium sulfate, and filtered. Distillation of the filtrate through a
75-cm. spinning-band column gives
133 g. (
68%) of
trans-2-butene oxide as a colorless oil, b.p.
52.5–55°.
3.
A slight excess of
trans-2-butene oxide is used to assure complete utilization of
methanesulfonic acid. The checkers' experiments indicated that a 15% excess of the epoxide substantially reduced the amount of unreacted methane–sulfonic acid present in the product and did not appear to interfere with the succeeding steps of this procedure.
4.
This order of addition and dilution is required to avoid dimerization or polymerization of the epoxide.
5.
No attempt was made to purify this compound further. It had a very characteristic
1H NMR spectrum (CDCl
3, external tetramethylsilane reference): δ 1.22 (d,
J = 7.5 Hz., 3H), 1.37 (d,
J = 7.5 Hz., 3H), 3.1 (s, 3H), 3.4 (s, O
H, position variable), 4.0 (d of q,
J = 4.0, 7.5 Hz., 1H), and 4.78 (d of q,
J = 4.0, 7.5 Hz., 1H). A sample stored for several weeks at room temperature showed no change in its spectrum.
6.
Cyclobutanecarboxylic acid chloride was obtained from Aldrich Chemical Company, Inc., and distilled prior to use. The acid chloride can be prepared by the reaction of
thionyl chloride with the corresponding acid (available from Aldrich) by the general procedure in
Org. Synth., Coll. Vol. 1, 147 (1941). The preparation of
cyclobutanecarboxylic acid has been described in
Org. Synth., Coll. Vol. 3, 213 (1955) and elsewhere.
27.
The
1H NMR spectrum (CDCl
3, external tetramethylsilane reference): δ 1.27 (d,
J = 6.5 Hz., 3H), 1.41 (d,
J = 6.5 Hz., 3H), 2.18 (m, 6H), 3.10 (s, 3H), superimposed on 3.2 (m, 1H)k, and 5.0 (m, 2H). IR (CDCl
3): 1725 cm.
−1.
8.
Commercial material from Matheson, Coleman and Bell and recrystallized reagent gave comparable results. The yield is decreased by use of less than
1 mole of sodium borohydride per mole of
mesylate.
9.
Either purified
pentane or Spectranalyzed
pentane available from Fisher Scientific Company was used.
10.
The submitter reports yields of
10–11 g. (
64–71%). The checker obtained the
dioxolane in
57% yield on conducting the experiment on a sixfold scale.
11.
The
1H NMR spectrum (neat, external tetramethylsilane reference) δ 1.1 (two overlapping d,
J = 6 Hz., 6H), 1.7–2.0 (m, 6H), 2.1–2.6 (m, 1H), 3.2–3.8 (m, 2H), and 4.94 (d,
J = 5 Hz., 1H).
12.
This proportion of water to
N,N-dimethylformamide is needed to assure solubility (hence facile reaction) of the
acetal on heating at reflux.
13.
The
1H NMR spectrum (neat, external tetramethylsilane reference): δ 1.4–2.4 (m, 6H), 2.6–3.2 (m, 1H), and 9.8 (d,
J ≈ 1.5 Hz., 1H); IR (CCl
4); 1730 cm.
−1 (C=O).
3. Discussion
Cyclobutanecarboxaldehyde has been prepared in very low yield by the Rosenmund reduction procedure.
3 A
46% yield of the 2,4-dinitrophenylhydrazone derivative has also been reported, with the aldehyde formed as an intermediate, in the reaction of the acid chloride and
lithium tri-t-butoxyaluminum hydride at −78° in diglyme.
4
Methods now available for the reduction of carboxylic acid derivatives to aldehydes require careful control of conditions to avoid overreduction or underreduction. The procedure described here is particularly convenient in that the acetal, not subject to further reduction, is formed directly in the reducing medium.
The scope of the reaction is indicated in Table I. An interesting aspect of the reaction is that the rate of the borohydride reduction step appears to be relatively insensitive to the substitutent R. It is suggested that the reaction occurs with formation of an intermediate acyloxonium ion, which is rapidly converted to acetal by reaction with the borohydride ion. Pyridine–borane has been shown to be the other product of this reaction; yield studies also indicate that only one hydride per borohydride ion is used efficiently in the formation of acetal.
TABLE I
ALDEHYDES FROM ESTER-MESYLATES
|
|
|
|
Ester-Mesylates (A), R =
|
Acetal (B) Yield, %a
|
Aldehyde (C) Yield, %a
|
|
|
n-C5H11-
|
63
|
78
|
|
C6H5-
|
64
|
89
|
|
cyclo-C6H11-
|
75
|
93
|
|
(CH3)3C-
|
77
|
50b
|
|
C6H5CH=CH-
|
62
|
87c,d
|
|
|
67
|
81c
|
|
cyclo-C3H5-
|
72
|
81
|
|
a Yield of distilled product; in several instances numerous runs were made, and the lowest yield is given.
|
b This acetal hydrolyzes quite slowly, and the relatively low yield of pivalaldehyde appears to be associated with this observation.
|
c Yields determined by GC analysis only.
|
d Fifteen percent of this product is the dihydro derivative, that is, the acetal of 3-phenylpropanal.
|
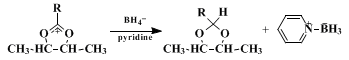
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
erythro-3-Methanesulfonyloxy-2-butyl cyclobutanecarboxylate
erythro-2,3-Butanediol monomesylate
erytho-2,3-butanediol monomesylate
acetal of 3-phenylpropanal
ACETAL (105-57-7)
potassium carbonate (584-08-7)
sulfuric acid (7664-93-9)
ether,
diethyl ether (60-29-7)
sodium acetate (127-09-3)
thionyl chloride (7719-09-7)
sodium hydrogen carbonate (144-55-8)
sodium chloride (7647-14-5)
sodium carbonate (497-19-8)
sodium sulfate (7757-82-6)
nitrogen (7727-37-9)
acetone (67-64-1)
pyridine (110-86-1)
Pentane (109-66-0)
magnesium sulfate (7487-88-9)
borane (7440-42-8)
N,N-dimethylformamide (68-12-2)
Cyclobutanecarboxylic acid (3721-95-7)
peracetic acid (79-21-0)
methanesulfonic acid,
mesylate (75-75-2)
sodium borohydride (16940-66-2)
1,1,2,2-tetrachloroethane (79-34-5)
Cyclobutanecarboxaldehyde (2987-17-9)
cyclobutanecarboxylic acid chloride (5006-22-4)
dioxolane (646-06-0)
pivalaldehyde (630-19-3)
lithium tri-t-butoxyaluminum hydride (17476-04-9)
trans-2-Butene (624-64-6)
trans-2-butene oxide (21490-63-1)
2-Cyclobutyl-cis-4-trans-5-dimethyl-1,3-dioxolane (36416-29-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved