Org. Synth. 1977, 57, 11
DOI: 10.15227/orgsyn.057.0011
ALDEHYDES FROM OLEFINS: CYCLOHEXANECARBOXALDEHYDE
Submitted by P. Pino
1 and C. Botteghi
2.
Checked by Mary M. Borecki, Joseph J. Mrowca, and Richard E. Benson.
1. Procedure
Caution! Benzene has been identified as a carcinogen; OSHA has issued emergency standards on its use. All procedures involving benzene should be carried out in a well-ventilated hood, and glove protection is required.
To a stainless-steel, 0.5-l. pressure vessel (Note 1) equipped with a 450-atm. manometer and a temperature recorder is added 0.2 g. (0.8 mmole) of rhodium(III) oxide (Note 2). The vessel is sealed and evacuated to 0.1 mm. pressure. A solution of 82 g. (1.0 mole) of cyclohexene (Note 3) in 140 ml. of anhydrous benzene is introduced by suction into the vessel. The vessel is placed in a heatable shaking device and pressured to 75 atm. with carbon monoxide; the total pressure is then increased to 150 atm. with hydrogen (Note 4). Shaking is begun and the vessel is heated to an internal temperature of 100° (Note 5). When the internal temperature reaches 100°, the pressure begins to fall. Whenever the pressure falls to 60 atm., rocking is stopped and the pressure is first increased to 105 atm. with carbon monoxide, then to 150 atm. with hydrogen. Rocking is started again, and the process is continued until no appreciable pressure decrease occurs. Approximately 2 hours is required, and the pressure decrease corresponds to the consumption of 2 moles of gas. The vessel is rapidly cooled to room temperature (Note 6) and the residual gas is carefully vented.
The vessel is opened, and the slightly yellow reaction mixture is transferred immediately to a 2-l., round-bottomed flask containing a freshly prepared solution of 200 g. of sodium hydrogen sulfite in 400 ml. of water. The flask is fitted with a stopper and is occasionally shaken at room temperature for a period of 3 hours (Note 7). The resulting precipitate is collected by suction filtration on a sintered-glass funnel and washed with 500 ml. of diethyl ether (Note 8). After drying in air, the bisulfite derivative is transferred to a 2-l. distillation flask containing 1 l. of 20% aqueous potassium carbonate. The resulting mixture is distilled, and the azeotropic mixture of water and aldehyde (b.p. 94–95°) is collected under nitrogen (Note 9).
The aldehyde is separated from the lower aqueous layer as a colorless liquid, dried over 10 g. of anhydrous sodium sulfate, filtered, and distilled under reduced pressure using a Claisen distillation apparatus, yielding 92–94 g. (82–84%) of cyclohexanecarboxaldehyde, b.p. 52–53° (18 mm.), nD25 1.4484 (Note 10), (Note 11). A purity of about 98% was established by GC analysis (Note 12); the product is suitable for synthetic use without further purification (Note 13).
2. Notes
1.
The pressure vessel was tested to a pressure of 700 atm. at 300°.
2.
The submitters used
rhodium(III) oxide available from Fluka A G without further purification. The checkers obtained
rhodium(III) oxide from Alfa Inorganics.
3.
The
cyclohexene was purified by distillation over
sodium metal before use (
nD25 1.4452). The submitters used the product available from Fluka A G, and the checkers used the product available from Aldrich Chemical Company, Inc.
4.
The purity of the gases used was greater than 99%.
5.
During the course of the reaction the temperature was maintained at 100° ± 2°.
6.
This procedure avoids secondary reactions of the aldehydes, which lead to high-boiling products. It is particularly advisable when linear aliphatic aldehydes are synthesized using
cobalt catalysts.
37.
The formation of the bisulfite derivative is an exothermic reaction; the flask is cooled with a bath of cold water for the first 10–15 minutes.
8.
It is impossible to obtain a completely white precipitate by this procedure.
9.
In order to avoid oxidation of the product the submitters recommend use of a
nitrogen atmosphere for all manipulations involving
cyclohexanecarboxaldehyde.
10.
Literature
4 values for
cyclohexanecarboxaldehyde: b.p.
78.5–80° (57 mm.),
nD25 1.4485.
11.
Cyclohexanecarboxaldehyde is stable at room temperature under
nitrogen; the submitters noted no appreciable variation in the refractive index after 30 days.
12.
The submitters state that GC analysis was made using a
2-m. column packed with polypropylene glycol (LB-550-X available from Perkin-Elmer) on Chromosorb. The retention time at 140° is 5.2 minutes at a flow rate of 30 ml./minute of
nitrogen. The 2,4-dinitrophenylhydrazone derivative
4 melts at 173–174°, and the semicarbazone derivative
5 melts at 172–173°.
13.
In addition to
rhodium(III) oxide,
cobalt(II) acetylacetonate or
dicobalt octacarbonyl has been used by the submitters as catalyst precursors for the hydroformylation of
cyclohexene. The results are given in Table I.
TABLE I
HYDROFORMYLATION OF CYCLOHEXENE WITH COBALT CATALYSTSa
|
|
Catalyst Precursor (mole/l.)
|
Solventb
|
Reaction Temperature
|
Reaction Time (hours)
|
Yieldc (%)
|
|
|
[Bis(acetylacetonate) cobalt(II)] (0.08)
|
Benzene
|
150°d
|
1.5
|
70
|
|
[Bis(acetylacetonate)e cobalt(II)] (0.08)
|
Heptane
|
110°
|
12
|
74
|
|
Dicobalt octacarbonylf (0.006)
|
Benzene
|
120°
|
8
|
80
|
|
a 4.15 mole/l. of cyclohexene; CO:H2 = 1:1; 150 atm. initial pressure.
|
|
|
c The aldehyde was isolated from the reaction mixture through its bisulfite derivative as described in the procedure.
|
d Induction time, 40–60 minutes.
|
e This catalyst precursor (5 g.) in 140 ml. of heptane was heated in the autoclave at 160° with a mixture of CO:H2 (1:1) at 150 atm. for 2 hours. The vessel was cooled, the gas released, 1 mole of cyclohexene was charged, and the reaction was carried out according to the usual procedure.
|
f The submitters used product available from Fluka A G that was dried under reduced pressure after recrystallization from heptane at −70°.
|
3. Discussion
This preparation is an illustration of the hydroformylation of olefins (oxo synthesis). The reaction occurs in the presence of soluble catalytic complexes of Group VIII metals. Although the metal originally used by Roelen
6 and still largely used in industry for the production of aliphatic aldehydes and alcohols
7 is
cobalt, the most active and selective catalysts are rhodium-containing compounds. The catalytic activity of the other Group VIII metals is, in general, much poorer. Although the hydroformylation of unsaturated substrates is a very general reaction,
7,8 some important limitations associated with the olefin structure may lead to the formation of isomeric aldehydes. In addition, especially in the presence of
cobalt catalysts, further reactions of synthesized aldehydes may occur under hydroformylation conditions.
With regard to the structure of the olefins, tetrasubstituted olefins do not undergo hydroformylation under typical reaction conditions, and olefinic substrates containing functional groups sometimes give poor yields and unexpected products.
7,8 If there is no plane of symmetry in the substrate across the double bond, at least two isomeric aldehydes are obtained.
9 Although methods for shifting the isomeric composition of the products have been proposed,
12,13,14 complete control of the isomeric composition has not been achieved despite the fact that the reaction mechanism is fairly well understood.
15 In addition, if the structure of the olefin is such that a double-bond shift is possible, isomers other than the two shown below can be formed.
10 Further reactions of the synthesized aldehydes may occur, especially when
cobalt catalysts are used, leading to alcohols, aldol condensation products or acetal derivatives. Some of the secondary reactions can be avoided by carrying out the hydroformylation in the presence of orthoformic acid esters
16 or of other reagents protecting the aldehyde group.
17 However, care must be taken when ortho esters are used, since hydroformylation of ortho esters may occur and yield aldehydes or acetals.
18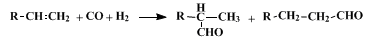
Although
cobalt catalysts are the best known and the most commonly used, in recent years
rhodium has been preferred for laboratory syntheses because of its higher activity and selectivity. As catalyst precursors Rh
2O
3,
11 Rh
4(CO)
12,
19 or HRh(CO)(PPh
3)
313 are commonly used.
Rhodium complexes supported on polymers have also been used.
14 For typical organic syntheses the easily accessible Rh
2O
3 seems preferable, even if higher temperature and pressures are required to carry out the olefin hydroformylation. Using HRh(CO)(PPh
3)
3, olefin hydroformylation at room temperature and pressure is possible.
13 Carrying out the reaction in the presence of
(−)DIOP [(4R,5R)-2,2-dimethyl-4,5-bis(5-dibenzophosphol-5-ylmethyl)-1,3-dioxolane], produced optically active aldehydes from monosubstituted ethylenes, as well as from 1,1- and 1,2-disubstituted ethylenes.
20The hydroformylation of
cyclohexene has been extensively investigated.
13,14,16,21,22,23 The present procedure is an adaptation of the
rhodium-catalyzed hydroformylation of
2-butene.
11Other methods for the preparation of
cyclohexanecarboxaldehyde include the catalytic hydrogenation of
3-cyclohexene-1-carboxaldehyde, available from the Diels–Alder reaction of
butadiene and
acrolein,
24 the reduction of
cyclohexanecarbonyl chloride by
lithium tri-tert-butoxyaluminum hydride,
25 the reduction of
N,N-dimethylcyclohexanecarboxamide with
lithium diethoxyaluminum hydride,
26 and the oxidation of the
methane sulfonate of cyclohexylmethanol with
dimethyl sulfoxide.
27 The hydrolysis, with simultaneous decarboxylation and rearrangement, of glycidic esters derived from
cyclohexanone gives
cyclohexanecarboxaldehyde.
4,28
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
cobalt(II) acetylacetonate
Bis(acetylacetonate) cobalt(II)
Bis(acetylacetonate)e cobalt(II)
(−)DIOP [(4R,5R)-2,2-dimethyl-4,5-bis(5-dibenzophosphol-5-ylmethyl)-1,3-dioxolane]
methane sulfonate of cyclohexylmethanol
potassium carbonate (584-08-7)
Benzene (71-43-2)
diethyl ether (60-29-7)
hydrogen (1333-74-0)
carbon monoxide (630-08-0)
Acrolein (107-02-8)
Cyclohexanone (108-94-1)
Cyclohexene (110-83-8)
sodium sulfate (7757-82-6)
nitrogen (7727-37-9)
sodium hydrogen sulfite (7631-90-5)
sodium (13966-32-0)
butadiene (106-99-0)
heptane (142-82-5)
cobalt (7440-48-4)
dimethyl sulfoxide (67-68-5)
N,N-Dimethylcyclohexanecarboxamide (17566-51-7)
Cyclohexanecarboxaldehyde (2043-61-0)
cyclohexanecarbonyl chloride (2719-27-9)
rhodium (7440-16-6)
rhodium(III) oxide (12036-35-0)
2-butene
3-cyclohexene-1-carboxaldehyde
lithium diethoxyaluminum hydride
lithium tri-tert-butoxyaluminum hydride (17476-04-9)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved