Org. Synth. 1986, 64, 1
DOI: 10.15227/orgsyn.064.0001
ADDITION OF AN ETHYLCOPPER COMPLEX TO 1-OCTYNE: (E)-5-ETHYL-1,4-UNDECADIENE
[1,4-Undecadiene, 5-ethyl-, (E)-]
Submitted by Ramnath S. Iyer and Paul Helquist
1.
Checked by Brian H. Johnston and Andrew S. Kende.
Checked by Ronald C. Newbold and Andrew S. Kende.
1. Procedure
Caution! This experiment should be performed in an efficient fume hood because of the unpleasant odor of dimethyl sulfide.
A dry, 1-L, one-necked, round-bottomed flask is equipped with a Teflon-coated magnetic stirring bar and a three-way stopcock bearing a rubber septum (Note 1), and the flask is charged with 25.2 g (0.123 mol) of the dimethyl sulfide complex of cuprous bromide (Note 2). An argon or nitrogen (Note 3) atmosphere is established in the flask by repeated cycles of evacuation with an oil pump and refilling with the inert gas. Through use of a syringe or cannula, 150 mL of diethyl ether (Note 4) and 120 mL of dimethyl sulfide (Note 4)) are added. After the mixture is stirred for a few minutes at 25°C, the resulting clear and colorless solution is cooled to−45°C (Note 5) and (Note 6). A 2.73 M solution (45.0 mL, 0.123 mol) of ethylmagnesium bromide in ether (Note 7) is added dropwise with a syringe or cannula over a period of 10 min. The suspension of yellow–orange solid is stirred at−45°C for 2 hr, and 1-octyne (16.0 mL, 0.109 mol; (Note 4)) is added with a syringe or cannula over a period of 2 min. After the solution is stirred at −45°C for 2 hr, it is cooled to −78°C (Note 5) and maintained at this temperature during the successive additions of N,N'-dimethylpropyleneurea, DMPU (27.7 mL, 0.223 mol; (Note 8)) and allyl bromide (11.4 mL, 0.131 mol; (Note 4)). The mixture is immediately warmed to −30°C and stirred at−30°C for 12 hr; it is warmed to 0°C and quenched by the addition of 30 mL of a saturated, aqueous solution of ammonium chloride adjusted to pH 8 with ammonium hydroxide. The mixture is stirred at 25°C in the air for 1.5 hr (Note 9) and is then shaken in a separatory funnel with a mixture of additional diethyl ether (50 mL) and water (50 mL). The dark-blue aqueous layer is drawn off and the organic layer is washed with additional 50-mL portions of the ammonium chloride solution (pH 8) until the washings are colorless. The organic layer is washed separately with water (50 mL) and saturated aqueous sodium chloride solution (50 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated by rotary evaporation at 25°C (25 min). The residue consists of 15.9 g of yellow oil that is purified by distillation under reduced pressure through a 15-cm Vigreux column to give 13.9 g (71%) (Note 10) of (E)-5-ethyl-1,4-undecadiene as a clear, colorless liquid, bp 56°C (0.70 mm; (Note 11)).
2. Notes
1.
The stopcock is constructed as shown below so that a source of inert gas and vacuum may be attached to the horizontal tubulation and liquid reagents and solutions may be transferred into the reaction flask with a syringe needle or cannula inserted through a rubber septum placed over the end of the vertical tubulation. In order to avoid air leaks through the septum into the reaction flask when reagents are not being added, the stopcock is normally turned to close off the vertical tubulation, but to leave the flask open to the argon source.
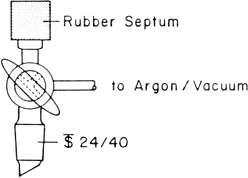
2.
This complex is prepared from
cuprous bromide and
dimethyl sulfide according to the procedure of House.
2 The complex must be pure white. Slightly impure samples will produce pinkish solutions that are unsatisfactory for this procedure. Normally, the complex is dark red when it is first prepared, but the required state of purity can be achieved by two or three recrystallizations under a
nitrogen atmosphere as described by House.
2 We have found, however, that if the initially formed complex has a distinctly green appearance, it cannot be purified satisfactorily. Others have also been concerned about this matter of purification.
3 43.
The checkers found that the yields were diminished when the reaction was run under prepurified
nitrogen, but they did obtain the reported yields using
argon. The submitters, however, have never experienced difficulty using the prepurified
nitrogen available in their laboratory.
4.
Commercially obtained materials were purified before use as described below.
Diethyl ether was distilled from a dark-blue or dark-purple solution of
sodium benzophenone radical anion or dianion under
nitrogen. This solution was obtained by dissolving
10 g of benzophenone in
1 L of commercial anhydrous ether, adding
10 g of freshly pressed sodium wire, and heating the mixture at reflux under
nitrogen until the characteristic blue or purple color developed.
Dimethyl sulfide (Aldrich Chemical Company, Inc.),
1-octyne (Chemical Samples Company or Albany International Chemicals), and
allyl bromide (Columbia Organics) were distilled under
nitrogen at atmospheric pressure.
Dimethylpropyleneurea (DMPU) (Aldrich Chemical Company, Inc.) was distilled at aspirator pressure from
calcium hydride.
5.
Constant temperatures were maintained by using
dry ice–acetone (−78°C) or dry ice–acetone/carbon tetrachloride baths (−25 to−45°C; the temperature tends toward the upper part of this range as the amount of
acetone used is decreased) or more conveniently through the use of an acetone bath equipped with a Neslab CryoCool Model CC-100F low-temperature unit, a Cole-Parmer Versa-Therm Model 2158 temperature controller, and a 500-W immersible heating coil. The temperature of the alkenylcopper solution must not be allowed to exceed−15°C; above this temperature rapid coupling to give a diene occurs.
6.
When the solution of the cuprous bromide complex is cooled, a portion of the reagent may precipitate, but this behavior does not affect the overall results of the experiment.
7.
The
ethylmagnesium bromide solution was obtained from Alfa Products, Morton Thiokol, Inc. and was titrated before use by the method of Watson and Eastham.
58.
(a) Checkers observed the freezing of some material on addition of DMPU and
allyl bromide, making it difficult to maintain stirring. Vigorous, frequent shaking was performed to ensure good mixing. (b) The submitters used
hexamethylphosphoric triamide, HMPA, in the original submission, but the Board of Editors of
Organic Syntheses decided that, in view of the suspected carcinogenicity of this solvent, all procedures using it would be rechecked with DMPU. See
(Note 10) for the effect of this substitution on the yield.
9.
Stirring the mixture in the air simplifies the workup procedure because cuprous complexes are oxidized to cupric compounds that are highly soluble in water or the aqueous
ammonia workup medium of this experiment.
10.
This yield is approximately 14% lower than that reported using HMPA; however, this result is consistent with those reported by Seebach
6 for the substitution of DMPU.
11.
The spectral characteristics of the final product are as follows: IR (neat) cm
−1: 3080, 2960, 2915, 2800, 1660, 1640, 1465, 1175, 940, and 910; H
1 NMR (80 MHz, CDCl
3) δ: 0.78–2.13 (several overlapping m, 18 H, other saturated C-H's), 2.63 (t, 2 H,
J = 6.5, C=CH-C
H2-C
H=CH
2), 4.70–5.20 (m, 3 H, other alkenyl C-H's), 5.50–6.05 (m, 1 H, alkenyl C-H); mass spectrum (70 eV)
m/e (relative intensity) 181.1 (M + 1, 1.3), 180.1 (M, 9.2).
3. Discussion
The procedure described above provides an approach to trisubstituted alkenes, compounds that are very common among natural products and that serve as key intermediates in the synthesis of other types of compounds.
7 The methods that have been developed for the preparation of trisubstituted alkenes are far too numerous to discuss to any significant extent here, but they have been the subject of previous review articles.
9 10 11 12 Very briefly, however, a large portion of the available methods may be divided among the following categories:
8 (1) elimination or cleavage reactions of organic halides and other compounds bearing leaving groups; (2) carbonyl condensation reactions of phosphonium ylides and other carbanionic or at least nucleophilic organic intermediates; (3) cleavage or rearrangements of other systems; (4) substitution reactions of alkenyl halides and related compounds; (5) reactions of various allylmetal and other allylic systems; (6) reactions of various alkenylmetal species; and (7) addition reactions to acetylenes, 1,3-dienes, and allenes.
The reaction of organocopper reagents with simple, unactivated acetylenes, an example of the last class of methods in the preceding list, serves as the basis of the procedure described here. This addition reaction was first reported by Normant in 1971
13 and has been investigated extensively since that time by not only Normant,
14 but also by Vermeer,
15 Helquist,
8 Levy,
16 and others.
4,
17 18 19 20 21 22 23 24 25 26 27 28 29 30,31 An important modification
8 of the reaction that has been incorporated in the present preparation is the use of
dimethyl sulfide as a ligand and cosolvent, which permits much higher yields and a broader range of applicability than the originally reported procedure.
13The specific example reported here is one of several preparations that have been developed using the same general reaction sequence. A brief summary of other representative cases is given in Table I.
8,
32 Notice that the alkenylcopper intermediates react with a variety of electrophilic reagents in addition to alkyl halides. It is especially
TABLE I
TRISUBSTITUTED ALKENES FROM ADDITION OF GRIGNARD-DERIVED ALKYLCOPPER COMPLEXES TO ACETYLENES, FOLLOWED BY REACTION WITH ELECTROPHILIC REAGENTS
|
|
Grignard Reagent
|
Acetylene
|
Electrophile
|
Product
|
Overall Yield(%)a,b
|
|
|
MeMgBr
|
|
|
|
84
|
|
MeMgBr
|
|
|
|
65
|
|
EtMgBr
|
|
|
|
94
|
|
|
|
CO2
|
|
50
|
|
|
|
|
|
73
|
|
aSince yields are sensitive to traces of oxygen during the reactions, the use of an argon atmosphere is strongly recommended.
|
bSee 9,10,11,12,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31.
|
noteworthy that the overall sequences leading to trisubstituted alkenes have been shown to proceed with greater than 99.9% stereoselectivity.
8 This unusually high degree of control of alkene configuration is of great value in natural products synthesis. The overall stereochemistry is indicative of
syn addition of the alkylcopper complexes to acetylenes, a result which is observed for several types of carbometallation (or insertion) reactions.
14In summary, the procedure described in this chapter is representative of a very general, highly stereoselective approach to trisubstituted alkenes. The usefulness of this methodology has already been demonstrated in total synthesis.
8,
21,
22,
24,
27,
29,
30,
31,
32
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
dimethyl sulfide complex of cuprous bromide
sodium benzophenone
acetylene (74-86-2)
ammonia (7664-41-7)
ether,
diethyl ether (60-29-7)
ammonium chloride (12125-02-9)
sodium chloride (7647-14-5)
Allyl bromide (106-95-6)
oxygen (7782-44-7)
nitrogen (7727-37-9)
acetone (67-64-1)
Benzophenone (119-61-9)
sodium wire (13966-32-0)
cuprous bromide (7787-70-4)
ammonium hydroxide (1336-21-6)
ethylmagnesium bromide (925-90-6)
magnesium sulfate (7487-88-9)
dimethyl sulfide (75-18-3)
argon (7440-37-1)
calcium hydride (7789-78-8)
hexamethylphosphoric triamide (680-31-9)
1-OCTYNE (629-05-0)
dimethylpropyleneurea,
N,N'-dimethylpropyleneurea (7226-23-5)
(E)-5-Ethyl-1,4-undecadiene,
1,4-Undecadiene, 5-ethyl-, (E)- (107445-19-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved