Org. Synth. 1990, 69, 80
DOI: 10.15227/orgsyn.069.0080
MIXED HIGHER-ORDER CYANOCUPRATE-INDUCED EPOXIDE OPENINGS: 1-BENZYLOXY-4-PENTEN-2-OL
[4-Penten-2-ol, 1-(phenylmethoxy)-]
Submitted by Bruce H. Lipshutz
1, Robert Moretti, and Robert Crow.
Checked by Gary L. Bolton, Steven G. Toske, and James D. White.
1. Procedure
A 100-mL, two-necked, round-bottomed flask (Note 1) equipped with a stirring bar and a rubber septum is evacuated and flame-dried under vacuum, then flushed with dry argon. This process is repeated 3 times. Anhydrous tetrahydrofuran (36 mL, (Note 2)) and distilled thiophene (3.05 g, 2.91 mL, 36.3 mmol, (Note 3)) are injected via syringe and the resulting solution is cooled to −78°C. Butyllithium in hexanes (12.8 mL, 2.83 M, 36.3 mmol, (Note 4)) is added dropwise via syringe. The resulting light-yellow solution is warmed to −20°C using a solid dry ice–carbon tetrachloride bath and stirred for 20 min.
A 500-mL, two-necked, round-bottomed flask equipped with a stirring bar and a rubber septum is charged with copper(I) cyanide (2.95 g, 33.0 mmol, (Note 5)). The flask is evacuated and gently flame-dried under vacuum (Note 6), then flushed with dry argon. The process is repeated 3 times. Anhydrous tetrahydrofuran (33 mL) is injected and the resulting slurry is cooled to −78°C. At this time, the previously prepared solution of 2-lithiothiophene in tetrahydrofuran (at −20°C) is added via cannula to the stirring slurry. At the end of the addition, the acetone–dry ice bath is exchanged for an ice bath. After 5 min (Note 7), the flask is again placed in a dry ice–acetone bath. Vinyllithium in tetrahydrofuran (16.7 mL, 1.98 M, 33.0 mmol, (Note 8)) is added dropwise after 15 min. Then the −78°C bath is exchanged for an ice bath. After 5 min, the reaction mixture is recooled to −78°C and a cooled (−20°C) solution of benzyl 2,3-epoxypropyl ether (4.93 g, 30.0 mmol, (Note 9)) in anhydrous tetrahydrofuran (30 mL) is added to the cuprate solution via cannula over a 10-min period. The reaction mixture is warmed to 0°C (Note 10). After 3 hr at 0°C, it is warmed to ambient temperature and stirred for an additional 1 hr. It is then cooled to −78°C and poured on to a solution of saturated aqueous ammonium chloride (135 mL) and concentrated aqueous ammonium hydroxide (15 mL). The mixture is stirred for an additional 15 min while the temperature of the system is allowed to rise. The mixture is filtered through Celite. The flask and the filter cake are rinsed with tetrahydrofuran (2 × 20 mL). The tetrahydrofuran is evaporated using a rotary evaporator, and the resulting aqueous layer is extracted with ethyl acetate (2 × 150 mL). Each organic layer is washed with water (75 mL) and brine (75 mL). The combined organic layers are dried over anhydrous sodium sulfate and concentrated with a rotary evaporator. The residue is purified by column chromatography on silica gel (Note 11), using a 6 : 1 mixture of petroleum ether and ethyl acetate as eluant (Note 12), to afford an oil (5.7 g, 29.6 mmol, 99%), which is distilled through a short-path distillation apparatus to give 4.20 g of an oil (26.1 mmol, 73%) (Note 13) as a colorless liquid, bp 85°C at 0.1 mm; IR (neat) cm−1: 3400, 3070, 3030, 1640, 1100, 740, 700; 1H NMR (CDCl3) δ: 2.26 (t, 2 H, J = 6.9), 2.41 (d, 1 H, J = 3.3), 3.4–3.6 (m, 2 H), 3.90 (m, 1 H), 4.55 (s, 2 H), 5.1 (m, 2 H), 5.75–5.90 (m, 1 H), 7.33 (s, 5 H); mass spectrum, m/e (relative intensity): 192 (M+, 1.06), 92 (24.89), 91 (100); high-resolution mass spectrum. Calcd. for C12H16O2: 192.1150. Found: 192.1161.
2. Notes
1.
All glassware, needles, and syringes are stored in an oven at 120°C overnight and assembled while hot.
2.
Tetrahydrofuran is distilled from
sodium–benzophenone before use.
3.
Thiophene is purchased from the Aldrich Chemical Company, Inc. and distilled from
calcium hydride before use.
4.
Butyllithium in hexanes (2.5 M) is purchased from the Aldrich Chemical Company, Inc. and titrated with
2-pentanol in ether, using
1,10-phenanthroline as indicator before use.
2 Use of lower concentrations of
butyllithium for the metalation of
thiophene under these conditions results in incomplete lithiation.
5.
Copper(I) cyanide is purchased from the Aldrich Chemical Company, Inc. and is dried in an Abderhalden apparatus at 56°C for ca. 2 days before use.
Caution: Copper(I) cyanide is very toxic.6.
Caution should be exercised during this operation. Overheating can result in partial decomposition of the copper(I) cyanide.7.
At this point, all of the
copper(I) cyanide has been consumed and the reaction appears as a brown solution.
8.
Vinyllithium in
tetrahydrofuran is purchased from Organometallics, Inc., and titrated
2 before use (see
(Note 4)).
9.
The starting material was prepared by the benzylation of
glycidol, with
benzyl bromide in
tetrahydrofuran according to the following procedure. A 500-mL, two-necked, round-bottomed flask, equipped with a stirring bar and a rubber septum, is evacuated and flame-dried under vacuum, then flushed with dry
argon. This process is repeated 3 times. A solution of distilled
glycidol (4.0 g, 3.58 mL, 54 mmol, obtained from Aldrich Chemical Company, Inc. and distilled before use) in anhydrous
tetrahydrofuran (139 mL) is injected via syringe and the resulting clear solution is cooled to 0°C.
Sodium hydride (2.24 g of a 60% dispersion in mineral oil, 56 mmol, obtained from Aldrich Chemical Company, Inc.) is added portionwise and the resulting gray mixture is stirred at 0°C for 30 min. Solid
tetrabutylammonium iodide (0.21 g, 0.56 mmol, obtained from Aldrich Chemical Company, Inc.) is then introduced all at once. Distilled
benzyl bromide (9.54 g, 6.63 mL, 55.8 mmol) is added (neat) dropwise via syringe. [
Benzyl bromide was purchased from the Aldrich Chemical Company, Inc. and dried over MgSO
4, filtered, and distilled before use. (
Caution: Benzyl bromide is light-sensitive and is a potent lachrymator.)] The solution is stirred at 0°C for 5 min, then warmed to ambient temperature and stirred for 1 hr. The mixture is quenched with aqueous
ammonium chloride and extracted with
ethyl acetate (2 × 200 mL). Each organic phase is washed with water (100 mL) and
brine (2 × 100 mL). The combined organic layers are dried over
sodium sulfate and concentrated using a rotary evaporator. The remaining yellow residue is purified by flash chromatography,
3 using a 5 : 1 mixture of
petroleum ether and
ethyl acetate as eluent, to afford chromatographically pure
(±)-benzyl 2,3-epoxypropyl ether (7.34 g, 44.7 mmol, 83%), which can be distilled through a short-path distillation apparatus to give
6.52 g of the epoxide (
39.7 mmol,
74%) as a colorless liquid, bp
105°C at 0.4 mm. Optically active
(S)-benzyl 2,3-epoxypropyl ether can be obtained in quantity in seven steps from
D-mannitol according to literature procedures.
4 Modified routes to
D-glyceraldehyde acetonide in bulk are also available,
5 as are other experimental changes for yield enhancements.
6 Reviews on the use of both
(R)/(S)-2,3-O-isopropylidene glyceraldehyde7 and nonracemic
glycidol and derivatives
8 provide excellent sources of additional information.
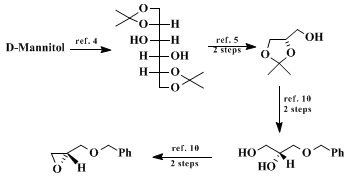
10.
At this point, the color of the solution turns from brown to light green, and darkens with time.
11.
The technique of Still
3 (flash chromatography) is used, with silica gel purchased from ICN Biomedicals (ICN Silica 21-63).
12.
For TLC analyses, Merck silica gel F-254 TLC plates were used, with 1 : 1
petroleum ether–
ether as eluent. Visualization was effected by spraying with a
5% phosphomolybdic acid in
ethanol solution followed by heating at ca. 250°C on a hot plate.
(R)-1-Benzyloxy-4-penten-2-ol has an
Rf of ca. 0.35 in this solvent system.
13.
The checkers found that there was consistently
10–12% of
1-benzyloxyheptan-2-ol in the reaction mixture resulting from coupling of "residual"
butyllithium (which forms
N-Bu(Th)Cu(CN)Li
2) with the epoxide.
3. Discussion
This procedure is an illustration of the use of mixed, higher-order (H.O.) cyanocuprates containing a nontransferable or "dummy" ligand for epoxide opening.
9 H.O. cuprates of general stoichiometry
RT(2-thienyl)Cu(CN)Li
2 can also be used to effect substitution reactions with halides, as well as conjugate additions to unhindered α,β-unsaturated ketones (see Table I).
9
TABLE I
REACTIONS OF VARIOUS SUBSTRATES WITH (VINYL)(2-THIENYL)CU(CN)LI2
|
|
Substrate
|
Equiv. of cuprate
|
Conditions
|
Product
|
Yield(%)
|
|
|
|
1.20
|
THF/Et2O room temp. 4 hr
|
|
71a
|
|
|
1.50
|
THF, 31° 18 hr
|
|
67b
|
|
|
1.10
|
THF/Et2O 1.1 eq. BF3 −78°, 1 hr
|
|
98b
|
|
|
1.25
|
THF/Et2O 0°, 1 hr
|
|
90a
|
|
aIsolated yield of chromatographically pure material.
|
bBy quantitative GC analysis.
|
The lower order (L.O.) cyanocuprate (2-Th)Cu(CN)Li has an excellent shelf life, thereby providing a highly stable precursor to higher-order cuprates.
10 Tetrahydrofuran solutions of this L.O. cuprate are available commercially (from Aldrich Chemical Company, Inc.), thus allowing further simplification of this procedure.
The often greater reactivity of H.O. cuprates
11 12 as compared to their L.O. counterparts to exemplified by this method. When
(vinyl)2CuLi (from 2
vinyllithium + 1 Cul, a Gilman-type reagent)
13 was used for the same transformation, a yield of
73% was observed, along with recovered starting material.
14 The L.O. cyanocuprate
(vinyl)Cu(CN)Li gave only
11% of the desired product.
9 Significantly, in the procedure described only 1.1 equiv of H.O. cuprate are necessary for complete consumption of the epoxide.
The
(R)-1-benzyloxy-4-penten-2-ol produced using enantiomerically enriched starting material in this reaction is a useful precursor in the synthesis of the polyol section of the polyene macrolide antibiotic roflamycoin (Scheme 1). This molecule has an array of 1,3-secondary hydroxyl groups, assumed to bear an all-
syn relationship to each other, for which a synthetic strategy has been devised.
14,15,16 Thus, a reiterative two-step protocol involving epoxide opening with a H.O.
vinylcyanocuprate, followed by stereoselective homoallylic alcohol epoxidation, reforms the functionality (i.e., an epoxide) from which it was originally derived (Scheme 2).
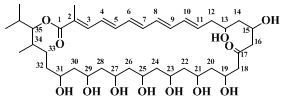

As an alternative to the preparation of (CH
2=CH-)(2-thienyl)Cu(CN)Li
2 from
vinyllithium (or a substituted
vinyllithium),
2-lithiothiophene, and CuCN, mixed reagents of this type can be formed via transmetallation processes involving vinylstannanes and
Me(2-thienyl)Cu(CN)Li2 in
THF at ambient temperatures.
17 Double-bond geometry is strictly maintained, and thus the intermediacy of vinylic organolithium species is avoided.
As additional development concerns the use of
vinylzirconium intermediates, formed in the usual manner by hydrozirconation of 1-alkynes. Treatment of these species at −78°C with
MeLi/Me2Cu(CN)Li2 effects ligand exchange, thereby forming a mixed cyanocuprate
(vinylic)MeCu(CN)Li2, which selectively delivers the vinyl residue to α,β-unsaturated ketones in high yields.
18,19 20 Alkylations can be effected using
MeLi/Me(2-thienyl)Cu(CN)Li2 to arrive at
(vinylic)(2-Th)Cu(CN)Li2.
21This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
petroleum ether
hexanes
brine
D-glyceraldehyde acetonide
(vinyl)2CuLi
(vinyl)Cu(CN)Li
Me(2-thienyl)Cu(CN)Li2
MeLi/Me2Cu(CN)Li2
(vinylic)MeCu(CN)Li2
MeLi/Me(2-thienyl)Cu(CN)Li2
(vinylic)(2-Th)Cu(CN)Li2
ethanol (64-17-5)
ethyl acetate (141-78-6)
ether (60-29-7)
ammonium chloride (12125-02-9)
sodium sulfate (7757-82-6)
copper(I) cyanide (544-92-3)
Benzophenone (119-61-9)
sodium (13966-32-0)
ammonium hydroxide (1336-21-6)
2-Pentanol (6032-29-7)
Thiophene (110-02-1)
benzyl bromide (100-39-0)
butyllithium (109-72-8)
Tetrahydrofuran,
THF (109-99-9)
sodium hydride (7646-69-7)
argon (7440-37-1)
calcium hydride (7789-78-8)
vinyllithium (917-57-7)
phosphomolybdic acid (51429-74-4)
1,10-phenanthroline (66-71-7)
2-lithiothiophene
1-Benzyloxy-4-penten-2-ol,
4-Penten-2-ol, 1-(phenylmethoxy)- (58931-16-1)
Benzyl 2,3-epoxypropyl ether,
(±)-benzyl 2,3-epoxypropyl ether (2930-05-4)
glycidol (556-52-5)
tetrabutylammonium iodide (311-28-4)
mannitol (69-65-8)
1-benzyloxyheptan-2-ol
vinylcyanocuprate
vinylzirconium
(R)/(S)-2,3-O-isopropylidene glyceraldehyde (15186-48-8)
(S)-benzyl 2,3-epoxypropyl ether (16495-13-9)
(R)-1-Benzyloxy-4-penten-2-ol
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved