Org. Synth. 1989, 67, 133
DOI: 10.15227/orgsyn.067.0133
N-BENZYL-N-METHOXYMETHYL-N-(TRIMETHYLSILYL)METHYLAMINE AS AN AZOMETHINE YLIDE EQUIVALENT: 2,6-DIOXO-1-PHENYL-4-BENZYL-1,4-DIAZABICYCLO[3.3.0]OCTANE
[Pyrrolo[3,4-c]pyrrole-1,3(2H,3aH)-dione, tetrahydro-2-phenyl-5-(phenylmethyl)-, cis-]
Submitted by Albert Padwa and William Dent
1.
Checked by Bruce Lefker and Albert I. Meyers.
1. Procedure
A. N-Benzyl-N-(trimethylsilyl)methylamine. An oven-dried, 100-mL, one-necked, round-bottomed flask equipped with a magnetic stirring bar and a reflux condenser is charged with 12.58 g (0.1 mol) of chloromethyltrimethylsilane (Note 1). Benzylamine (Note 2) (33.1 g, 0.3 mol) is added with stirring and the resulting solution is heated at 200°C for 2.5 hr. At the end of this time a 0.1 N sodium hydroxide solution is added in order to hydrolyze the white organic salt that had formed. The mixture is extracted with ether and the ether layer is dried over magnesium sulfate and concentrated under reduced pressure. The residue is distilled under reduced pressure through a 6-in. Vigreux column to give 11.6–15.3 g (58–72%) of N-benzyl-N-(trimethylsilyl)methylamine, bp 68–72°C (0.7–0.8 mm) (Note 3).
B. N-Benzyl-N-methoxymethyl-N-(trimethylsilyl)methylamine. A 25-mL, round-bottomed flask equipped with a stirring bar is charged with 6.0 g (74 mmol) of a 37% aqueous formaldehyde solution (Note 4). The solution is cooled to 0°C and 10.0 g (51.7 mmol) of N-benzyl-N-(trimethylsilyl)methylamine is added dropwise with stirring. After the solution is stirred for 10 min at 0°C, 6 mL (0.15 mol) of methanol (Note 5) is added in one portion. Potassium carbonate (4.0 g) is added to the mixture to absorb the aqueous phase. The mixture is stirred for 1 hr, the nonaqueous phase is decanted, an additional 2.0 g of potassium carbonate is added, and the mixture is stirred at 25°C for 12 hr. Ether is added to the mixture and the solution is dried over potassium carbonate, filtered, and concentrated under reduced pressure (Note 6). The residue is distilled at reduced pressure to give 6.8–8.6 g (54–69%) of N-benzyl-N-methoxymethyl-N-(trimethylsilyl)methylamine as a colorless liquid, bp 77–80°C (0.5 mm) (Note 7).
C. 2,6-Dioxo-1-phenyl-4-benzyl-1,4-diazabicyclo[3.3.0]octane. An oven-dried, 250-mL, one-necked, round-bottomed flask equipped with a magnetic stirring bar is charged with 10.0 g (0.042 mol) of N-benzyl-N-methoxymethyl-N-(trimethylsilyl)methylamine and 100 mL of anhydrous acetonitrile (Note 8). N-Phenylmaleimide (Note 9) (7.3 g, 0.042 mol) is added, followed by 1.7 g (0.063 mol) of lithium fluoride (Note 10). The reaction mixture is sonicated (Note 11) for 3 hr and poured into 100 mL of water. The mixture is extracted with three 100-mL portions of ether. The organic extracts are combined and washed with 100 mL of saturated sodium chloride solution, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The residue is chromatographed on a silica gel column (300 g) using a 35% ethyl acetate:hexane mixture (ca. 1500 mL) as the eluant to give 9.2–9.6 g (72–75%) of 2,6-dioxo-1-phenyl-4-benzyl-1,4-diazabicyclo[3.3.0]octane as a pale yellow solid, mp 97–98°C (Note 12).
2. Notes
1.
Chloromethyltrimethylsilane is purchased from Petrarch Systems, Inc. and is used without purification.
2.
Benzylamine, purchased from Aldrich Chemical Company, Inc., is distilled and stored over
potassium hydroxide.
3.
The submitters report bp
89–90°C (5 mm). The
1H NMR spectrum (CDCl
3, 90 MHz) is as follows δ: 0.10 (s, 9 H), 2.00 (s, 2 H), 3.78 (s, 2 H) and 7.28 (s, 5 H).
4.
Formaldehyde (37% solution in water) is purchased from Aldrich Chemical Company, Inc. Sufficient aqueous
10% sodium hydroxide solution (1–5 drops) is added until the pH reaches 7.
5.
Purified-grade methanol, purchased from Fisher Scientific Company, is used.
6.
The submitters found it easier to pump down the crude mixture overnight under reduced pressure to ensure that all the
methanol is removed. If not, the residue tends to bump uncontrollably upon distillation.
7.
The spectral properties are as follows: IR (neat) cm
−1: 3095, 3064, 3030, 2900, 1605, 1495, 1450, 1422, 1385, 1362, 1245, 1070, 925, 845, 740, 700;
1H NMR (CDCl
3, 90 MHz) δ: 0.10 (s, 9 H), 2.13 (s, 2 H), 3.20 (s, 3 H), 3.72 (s, 2 H), 3.95 (s, 2 H) and 7.22 (m, 5 H).
8.
Anhydrous acetonitrile, purchased from Aldrich Chemical Company, Inc., is distilled over
calcium hydride and stored over Linde 4A molecular sieves.
9.
N-Phenylmaleimide is purchased from Aldrich Chemical Company, Inc., and used without purification.
10.
Lithium fluoride is purchased from Fisher Scientific Company.
11.
A Branson ultrasonic cleaner (2.8 L, 13 × 23 × 10 cm), purchased from Fisher Scientific Company, is used for sonication. Without sonication, the yield drops by ca. 10–15%.
12.
The spectral properties are as follows: IR (neat) cm
−1: 3145, 3000, 2950, 2900, 2800, 1760, 1700, 1575, 1490, 1445, 1380, 1310, 1200, 1155, 880, 840, 740, 700;
1H NMR (CDCl
3, 90 MHz) δ: 2.3–2.7 (m, 2 H), 3.2–3.6 (m, 4 H), 3.60 (s, 2 H), 7.0–7.7 (m, 10 H).
3. Discussion
The preparation of pyrrolidines has received extensive attention by synthetic chemists in recent years, in part because of the interesting biological activities exhibited by several polysubstituted pyrrolidines.
2 Little attention has been given to one of the most conceptually simple ways of
pyrrolidine formation: a 1,3-dipolar cycloaddition of an azomethine ylide with an olefin. This is not surprising since few methods exist for the preparation of nonstabilized azomethine ylides.
3,4,5,6,7,8,9,10,11,12,13 Silyl-substituted amines of type 1 represent conjunctive reagents that can be considered as the equivalent of a nonstabilized azomethine ylide. These reagents have recently been found to undergo 1,3-dipolar cycloaddition to olefins to give pyrrolidine derivatives in good yield.
14,15,16 The present procedure provides a convenient route for the synthesis of a variety of five-membered ring nitrogen heterocycles using different dipolarophiles. Some representative examples are given in Table I. Advantages of the present method over existing methodologies include mild conditions, high yield, and simplicity of the cycloaddition.
Trimethylsilyl triflate or trimethylsilyl iodide can also be used.
12 However, these reagents are expensive and require longer reaction times.
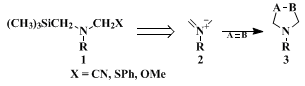
TABLE I
CYCLOADDITION OF 1 WITH ELECTRON-DEFICIENT DIPOLAROPHILES
|
|
Dipolarophile
|
Product
|
% Yield
|
|
|
|
|
90
|
|
|
|
90
|
|
|
|
90
|
|
|
|
92
|
|
|
|
80
|
|
|
|
91
|
|
We have found that sonication of the reaction mixture decreases the time needed for reaction and also substantially increases the yield. This is probably related to an increase in the solubility of lithium fluoride in acetonitrile or is a consequence of surface effects on the metal.
N-Benzyl-
N-methoxymethyl-
N-(trimethylsily)methylamine undergoes stereospecific cycloaddition with
dimethyl maleate and
fumarate. The cycloaddition behavior of an unsymmetrically substituted
α-methoxysilylamine has also been examined and found to occur with high overall regioselectivity. The stereospecificity and regioselectivity of the reaction is consistent with a concerted 1,3-dipolar cycloaddition reaction.
17
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
potassium carbonate (584-08-7)
ethyl acetate (141-78-6)
methanol (67-56-1)
ether (60-29-7)
acetonitrile (75-05-8)
sodium hydroxide (1310-73-2)
formaldehyde (50-00-0)
sodium chloride (7647-14-5)
potassium hydroxide (1310-58-3)
magnesium sulfate (7487-88-9)
benzylamine (100-46-9)
pyrrolidine (123-75-1)
hexane (110-54-3)
calcium hydride (7789-78-8)
Dimethyl fumarate (624-49-7)
N-Phenylmaleimide (941-69-5)
trimethylsilyl iodide (16029-98-4)
trimethylsilyl triflate (27607-77-8)
chloromethyltrimethylsilane (2344-80-1)
2,6-Dioxo-1-phenyl-4-benzyl-1,4-diazabicyclo[3.3.0]octane
lithium fluoride (7789-24-4)
dimethyl maleate (624-48-6)
α-methoxysilylamine
N-BENZYL-N-METHOXYMETHYL-N-(TRIMETHYLSILYL)METHYLAMINE (93102-05-7)
N-benzyl-N-(trimethylsilyl)methylamine
Pyrrolo[3,4-c]pyrrole-1,3(2H,3aH)-dione, tetrahydro-2-phenyl-5-(phenylmethyl)-, cis- (87813-00-1)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved