Org. Synth. 1990, 69, 19
DOI: 10.15227/orgsyn.069.0019
ENANTIOSELECTIVE SAPONIFICATION WITH PIG LIVER ESTERASE (PLE): (1S,2S,3R)-3-HYDROXY-2-NITROCYCLOHEXYL ACETATE
[1,3-Cyclohexanediol, 2-nitro-, 1-acetate, [1S-(1α, 2β, 3α]]
Submitted by Martin Eberle, Martin Missbach, and Dieter Seebach
1.
Checked by David L. Coffen
1. Procedure
A. (1R,2r,3S)-2-Nitrocyclohexane-1,3-diol. A 1-L, round-bottomed flask, equipped with a magnetic stirrer, a thermometer, and an ice–ethanol bath is charged with 175 mL (0.455 mol) of an aqueous 25% solution of glutaric dialdehyde, 38 mL (0.708 mol) of nitromethane, and 600 mL of methanol (Note 1). At 0–5°C 12 mL of aqueous 2 M sodium hydroxide is added gradually. The cooling bath is removed and the reaction mixture is stirred for 4 hr at room temperature. The resulting yellow solution is neutralized by adding 15 g of acidic cation-exchange resin and stirring for an additional 20 min (Note 2). The resin is filtered off and washed with a small volume of methanol. The filtrate is evaporated to a semisolid residue using reduced pressure and a 35°C water bath. The residue is dissolved in 100 mL of absolute ethyl alcohol with heating and diluted by gradual addition of 250 mL of toluene. The resulting two-phase mixture (Note 3) is again evaporated, with azeotropic removal of water. The resulting residue is again taken up in 100 mL of hot ethyl alcohol and diluted with 250 mL of toluene (Note 3). The almost colorless crystals are filtered and dried at high vacuum to yield 44–52 g (60–70%) of nitrodiol, mp 152–155°C (dec).
B. (1R,2r,3S)-3-Acetoxy-2-nitrocyclohexyl acetate. In a 1-L flask 52 g (0.323 mol) of the nitrodiol are suspended in 150 mL of acetic anhydride. Without cooling, 3–6 drops of concentrated sulfuric acid are added (Note 4). After 1 hr 500 mL of ice/water is rapidly added and stirring is continued for 60 min. The resulting colorless crystals are filtered, washed with water, and air-dried. The product thus obtained, 75.6 g (95.6%) of colorless crystals, is pure by TLC (4 : 1 hexane/ethyl acetate) and NMR, and melts at 89–90°C (Note 5).
C. (1S,2S,3R)-3-Hydroxy-2-nitrocyclohexyl acetate. A 500-mL flask equipped with a magnetic stirrer is charged with 10 g (41 mmol) of powdered nitrodiacetate and 300 mL of 0.2 M phosphate buffer of pH 7.0 prepared by dissolving 11 g of potassium dihydrogenphosphate and 3.3 g of potassium hydroxide in 300 mL of deionized (or distilled) water. To the stirred suspension is added 30 mg of purified PLE [Esterase (EC 3.1.1.1), suspended in 2.8 mL of 3.2 M ammonium sulfate buffer] (Note 6) and the mixture is stirred for 24 to 48 hr (Note 7). The continuously measured pH drops to about 5.6 during the reaction and then remains almost constant, while the mixture has turned to a practically clear, pale-yellow solution (Note 8). The solution is filtered through a paper filter and the filtrate is extracted 3 times with 100 mL of ether. The organic phase is dried over anhydrous magnesium sulfate and filtered. Removal of the solvent and of the acetic acid under reduced pressure leaves 7–8 g (85–95%) of colorless monoacetate as a crystalline solid (Note 9). Recrystallization by dissolving in 100 mL of ether and adding 250 mL of pentane gives 5–6 g (60–70%) of pure (1S,2S,3R)-3-hydroxy-2-nitrocyclohexyl acetate (Note 10), mp 90–91°C, [α]D + 9.5° (CHCl3, c 1.0) (Note 11). From the mother liquor, another 1–2 g (12–24%) of monoacetate, mp 89–90°C, can be obtained (Note 12).
2. Notes
1.
Commercial-grade chemicals were used without further purification. The
glutaric dialdehyde solution should be fresh.
2.
The checkers used Amberlite IR-120 (plus) acid form, capacity 1.9 meq/mL supplied by the Aldrich Chemical Company, Inc.
3.
Two layers will form unless the water content of the crude product is sufficiently reduced during the preceding evaporation; in that case, the evaporation is to be repeated.
4.
The solution turns clear and the temperature rises to 60–70°C when the reaction has started. More
sulfuric acid should be added if a sustained exotherm does not ensue.
5.
The submitters recommend recrystallization from alcohol/water (2 : 1). This is essential if NMR and TLC analyses of the crude nitrodiacetate indicate the presence of monoacetate.
6.
The checker used the contents of one 30-mg vial of Sigma material, rinsed in with ca. 1 mL of deionized water. The submitters originally developed the process with PLE purified according to a procedure they had published previously.
27.
The submitters used 45 mg of enzyme and observed the reaction to be complete in 11.5–12.5 hr.
8.
In addition to the monoacetate, a small amount of diacetate and an even smaller amount of diol could be detected by TLC in the crude product.
9.
The crude product is enantiomerically pure according to
19F NMR of the corresponding Mosher ester (>97% ee). The checker observed lower
[α]D25 values (+8.72° and +8.61°) but confirmed the enantiomeric purity by HPLC analysis of the corresponding Mosher esters. By HPLC comparison with the diastereomeric mixture of Mosher esters prepared from a sample of racemic monoacetate (oily substance obtained by partial hydrolysis of diacetate), the (−)-enantiomer content appears to be less than 1%.
10.
The absolute configuration of the product has been proved by X-ray analysis of the corresponding camphanic ester.
2,311.
The monoacetate shows the following
1H NMR spectrum (90 MHZ) δ: 1.3–1.9 (m, 4 H), 2.0 (s, 3 H), 2.1–2.2 (m, 2 H), 2.7 (d,
J = 5.5, OH), 4.1 (m, 1 H), 4.4 (apparent, t,
J = 10.5, 1H), 5.1–5.3 (m, 1 H).
12.
Recrystallization of this fraction gave another
0.75–1.5 g (
10–18%) of pure product.
3. Discussion
The use of ester-cleaving enzymes will probably be one of the most useful biological–chemical methods in the synthetic laboratory. No example of this type of reaction has hitherto been published in the
Organic Syntheses series of procedures. So far, the only biological–chemical
Organic Syntheses procedures are two yeast reductions,
4,5 one oxidation with horse liver alcohol dehydrogenase,
6 and a disaccharide synthesis catalyzed by emulsin.
7 The procedure described here is also applicable with crude enzyme powder, but the workup is a bit more complicated, because a continuous extractor must be used to overcome problems with emulsions. Both crude enzyme concentrate and purified PLE as a mixture of isoenzymes
8 are commercially available, but the crude concentrate can easily be prepared from fresh pig liver and is thus very cheap.
2 By using self-made PLE-powder, the submitters have produced amounts of
20–25 g of pure monoacetate per run.
2Several review articles containing discussions of enantioselective syntheses with ester-cleaving enzymes have appeared.
9,10,11,12,13 Of the many examples, the ones in which
meso substrates are employed are most attractive since the theoretical yield is 100%. In many applications of PLE the enantiomeric excess (% ee) of the product depends crucially on the source of the enzyme. This effect has not been noticed in the enantioselective saponifications of nitrodiol diacetates, either because the reaction is insensitive to it, or because this complication is overcome by the great crystallization tendency of the products. The only problem we observed when the reaction was carried out with commercial crude PLE-powder (but not with the self made one) was the production of a certain amount of diol that could not be removed by simple recrystallization. In this case, filtration over a
short silica gel column with
methylene chloride as eluent gives after recrystallization pure monoacetate.
Other examples of enantiomerically pure monoacetates of meso-nitrodiols, which are available using the procedure described above, are collected in Table I. Entries 1 and 3 in Table I refer to runs following the above procedure, for all other cases the self made crude PLE-powder was used. Cases in which no reaction (a) or unsatisfactory selectivities (b) were observed, are shown below.
TABLE I
YIELDS, MELTING POINTS, AND SPECIFIC ROTATIONS OF NITRODIOL MONOACETATES PREPARED BY PROCEDURE C USING PLE-POWDER2 INSTEAD OF PURIFIED ENZYMEa
|
|
Entry No.
|
|
Yield (%)
|
Mp (°C)
|
[α]D (CHCl3, c 1)
|
|
|
1
|
|
50–70
|
58–60
|
−10.5
|
|
2
|
|
40–50
|
Oil
|
−10.7
|
|
3
|
|
60–70
|
90–91
|
+9.8
|
|
4
|
|
80–90
|
106–107
|
−9.4
|
|
5
|
|
70–80
|
91–92
|
+29.1
|
|
6
|
|
60–70
|
130–131
|
−1.3
|
|
7
|
|
20–30
|
111–112
|
+14.7
|
|
8
|
|
60–70
|
130–131 (dec)
|
+28.1
|
|
9
|
|
60–70
|
93–94
|
+6.0
|
|
aEntries 1 and 3 refer to runs following the above procedure; for all other cases the self made crude PLE-powder was used. The configuration and the sense of chirality of the products of entries 1, 3, and 4 were determined by x-ray crystal structure analysis of the camphanic esters, those of the other are inferred by analogy and by NMR comparison. The open-chain compounds (entries 1 and 2) were obtained using TES buffer at pH 6.5.
|
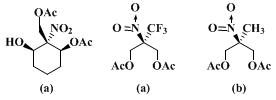
The
meso-nitrodiol starting materials for the preparation of the PLE diacetate substrates are readily obtained from
nitromethane or
nitroethane and aldehydes or dialdehydes. They crystallize readily. The above procedures for the preparation of
nitrocyclohexanediol and its diacetate from
glutaraldehyde and
nitromethane are modifications of published methods.
14,15The chiral monoacetates now available are useful multiple coupling reagents
16,17,18 for syntheses of enantiomerically pure target molecules. They can be converted to nitroolefinic allylic esters, achiral or racemic analogs of which we have previously shown
16,17,18 to combine sequentially with two (different) nucleophiles (see
1 and
2 in Scheme 1).
In a first approach to an enantioselective version of this method, we employed
19 chiral enamines derived from
proline,
20 (see
1–3 in Scheme 1). In this stoichiometric, enantioselective reaction, the valuable auxiliary used has to be recovered (i.e., recycled) in preparative-scale applications.
21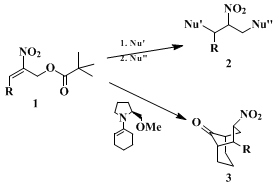
We then used
22 nitroallylic pivalates for the alkylation of hydroxy acid-derived enolates to prepare enantiomerically pure compounds (EPC), (see
4 and
5 in Scheme 2), an example of the use of the pool of chiral building blocks for EPC syntheses.
23,24,25 Finally, the procedure described here allows for syntheses of EPC with a catalytic enantioselective step:
26 dehydration of the monoacetate from the PLE saponification leads to
(S)-nitrocyclohexenyl acetate 6, and pivaloylation followed by acetate hydrolysis and dehydration leads to the
pivalate 8 of the enantiomeric alcohol. These compounds can be used for substitutions with a variety of nucleophiles.
2,3,26 Thus, starting from the enantiomerically pure Michael acceptors
6 and
8, 3-alkyl nitrocyclohexenes
7 9, respectively, of high enantiomeric excess are available (see Scheme 2, Nu =
methyllithium, phenyllithium and the morpholinoenamine of acetophenone):
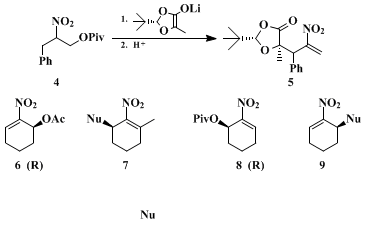
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
methyllithium, phenyllithium and the morpholinoenamine of acetophenone
1,3-Cyclohexanediol, 2-nitro-, 1-acetate, [1S-(1α, 2β, 3α]
ethyl alcohol (64-17-5)
sulfuric acid (7664-93-9)
acetic acid (64-19-7)
ethyl acetate (141-78-6)
methanol (67-56-1)
ether (60-29-7)
acetic anhydride (108-24-7)
sodium hydroxide (1310-73-2)
potassium hydroxide (1310-58-3)
toluene (108-88-3)
ammonium sulfate (7783-20-2)
Pentane (109-66-0)
Nitromethane (75-52-5)
methylene chloride (75-09-2)
magnesium sulfate (7487-88-9)
potassium dihydrogenphosphate (7778-77-0)
proline (147-85-3)
hexane (110-54-3)
glutaraldehyde,
glutaric dialdehyde (111-30-8)
nitroethane (79-24-3)
meso-nitrodiol
nitrocyclohexanediol
pivalate
(1S,2S,3R)-3-Hydroxy-2-nitrocyclohexyl acetate
(S)-nitrocyclohexenyl acetate
(1R,2r,3S)-2-Nitrocyclohexane-1,3-diol (38150-01-5)
(1R,2r,3S)-3-Acetoxy-2-nitrocyclohexyl acetate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved