Org. Synth. 1987, 65, 159
DOI: 10.15227/orgsyn.065.0159
ALLYLCARBAMATES BY THE AZA-ENE REACTION: METHYL N-(2-METHYL-2-BUTENYL)CARBAMATE
[Carbamic acid, (2-methyl-2-butenyl)-, methyl ester]
Submitted by Günter Kresze, Hans Braxmeier, and Heribert Münsterer
1.
Checked by N. Laxma Reddy and Ian Fleming.
1. Procedure
Caution! All three parts of this preparation should be performed in a well-ventilated hood. The reagents and the products of Parts A and B are toxic and unpleasant substances.
A. Methyl N,N-dichlorocarbamate. A 2-L, two-necked, round-bottomed flask equipped with a magnetic stirrer, a gas inlet, and a gas outlet is charged with 84 g (1.1 mol) of methyl carbamate, 210 g (2.6 mol) of sodium acetate, 21 g (0.35 mol) of glacial acetic acid, and 400 mL of water and cooled to −10 to −15°C (Note 1). About 175 g (2.5 mol) of chlorine is condensed in a calibrated Schlenk tube (Note 2) cooled with dry ice–methanol. The cooling bath is replaced by an ice–water bath (Note 3) and chlorine is passed slowly (Note 4) (over 2 hr, (Note 5)) at a constant rate into the solution, which is vigorously stirred with a magnetic stirrer. The mixture is transferred to a separatory funnel and the yellow oil which settles is run off. The yellow oil is washed with three subsequent 50-mL portions of a 20% aqueous sodium chloride solution and dried over anhydrous magnesium sulfate. The crude product is then transferred to a distillation apparatus, where it is kept under reduced pressure (11–15 mm) at room temperature for about 20 min. The bath temperature is slowly raised to a maximum of 60°C. The product distills at 43°C (11 mm) to give 102–118 g (63–73%, based on methyl carbamate) of a heavy yellow oil ((Note 6), (Note 7), (Note 8)).
B.
N1,N2-Bis(methoxycarbonyl)sulfur diimide. A
250-mL, three-necked, round-bottomed flask, equipped with a gas outlet stopcock, a
thermometer, and a
pressure-equalizing dropping funnel and containing a magnetic stirring bar, is purged with dry
nitrogen and charged with
53.0 g (0.37 mol) of methyl N,N-dichlorocarbamate and
0.2 mL of pyridine. The
dropping funnel is filled with
20.6 g (0.2 mol) of freshly distilled sulfur dichloride (Note 9). The whole apparatus is closed and connected by way of the gas outlet to a paraffin-oil-filled valve
(Note 10). About
2 mL of sulfur dichloride is then added dropwise into the stirred flask. When the evolution of
chlorine has started (usually after about 5 min)
(Note 11), the remainder of the
sulfur dichloride is added at such a rate that three to five bubbles of
chlorine per second are evolved
(Note 12), without allowing the temperature of the reaction mixture to exceed 35°C for more than short intervals. The addition takes 1.5–2 hr. After the addition is completed and the evolution of gas has slowed down significantly, volatile materials are removed by stirring the mixture at 60°C under reduced pressure (11–15 mm) for about 10 min
(Note 13). Further removal is accomplished at room temperature at 0.01–0.05 mm for approximately 1 hr. The product (
33.0–35.0 g), a moisture-sensitive, viscous, yellow oil, is used directly without further purification
(Note 14).
C.
Methyl N-(2-methyl-2-butenyl)carbamate. A
250-mL, two-necked flask, equipped with a gas outlet stopcock and a pressure-equalizing dropping funnel, and containing a magnetic stirring bar, is flushed with dry
nitrogen and charged with a solution of
N1,N2-bis(methoxycarbonyl)sulfur diimide from Part B in
30 mL of dry chloroform. The solution is cooled with ice water, and
14.1 g (0.20 mol) of 2-methyl-2-butene is slowly dropped in with stirring over 1 hr. After the addition is completed, the reaction mixture is stirred for another 10 hr at room temperature. The funnel is replaced by a distillation unit and most of the solvent is removed (11–15 mm, bath temperature 30°C). To the residue are added
240 mL of a 10% solution of potassium hydroxide in
methanol and about 10 mL of water. The mixture is stirred for 3 hr at room temperature, and any precipitate is filtered off
(Note 15). The residue is washed with
50 mL of methanol, and the filtrate, a red solution, is concentrated at 40°C (11–15 mm). The residue is taken up in
450 mL of ether and washed with four 100-mL portions of water. The ethereal layer is dried with anhydrous
magnesium sulfate and treated with charcoal until the color has changed from red to yellow, whereupon the solvent is removed under reduced pressure. The resultant oil is distilled at
70–75°C (3 mm), 62–64°C (0.2 mm) to give the colorless product, yield
10.7–13.7 g (
43–52%, based on
N,N-dichlorocarbamate)
(Note 16) and
(Note 17).
2. Notes
1.
The solution of
methyl carbamate,
sodium acetate, and
acetic acid is best prepared at room temperature. On cooling small amounts of precipitate form, but this precipitate dissolves again during the reaction.
2.
An amount of 175 g of condensed
chlorine corresponds to a volume of about 115 mL. A dry ice–acetone condenser is fitted above the Schlenk tube.
3.
A safety flask of at least 1 L should be placed between the Schlenk tube and the reaction flask.
4.
To prevent
chlorine from escaping before reaction has taken place, the gas inlet tube is immersed as deeply as possible into the solution.
5.
To follow the rate of evaporation of
chlorine, it is helpful to have calibrated the Schlenk tube.
6.
The crude product should not be stored, and the distilled product is best kept protected from light at −78°C.
7.
During the distillation, the product is best kept cooled in an
ice–water bath.
8.
The product is a powerful skin irritant. Protective gloves should be worn during the separation and when transferring the liquid to the distillation apparatus.9.
Sulfur dichloride decomposes at its boiling point. It is best distilled at low pressure, condensing it in a cooled vessel.
10.
The valve serves as a bubble counter for monitoring the evolution of gas.
11.
Sometimes no significant evolution of
chlorine occurs. In these cases the mixture is heated to 30–40°C with a
water bath, which is removed after the reaction has started. The checkers found no delay in the evolution of
chlorine.
12.
This evolution should not be interrupted.
13.
An aspirator and a water bath may be used for this purpose. A tube filled with anhydrous
calcium chloride should be placed between the aspirator and the flask.
14.
The product is best stored protected from light at −78°C. Even under these conditions, it is advisable to use it up within at least 1 month.
15.
Sometimes no significant amount of precipitate is formed, in which case filtration may be omitted.
16.
During distillation the receiving flask should be cooled with ice water in order to minimize losses. The product is a semisolid material at room temperature and solidifies completely when kept in a
refrigerator.
17.
The product has the following spectroscopic properties: IR (film) cm
−1: 3520 (NH), 1700 (CO) 1530;
1H NMR (250 MHz,
d5-pyridine) δ: 1.50 (d, 3 H,
J = 6.7, CH
3CH), 1.59 (s, 3 H, CH
3C=), 3.72 (s, 3 H, CH
3O), 3.90 (d, 2 H,
J = 6, CH
2N), 5.48 (q, 1 H,
J = 6.7, HC=), 8.00 (m, 1 H, HN), with long-range splitting evident in the fine structure. It appears to be a single stereoisomer (>97 : 3).
3. Discussion
The preparation of
methyl N,N-dichlorocarbamate is based on work of Foglia and Swern,
2 and the preparation of the
sulfurdiimide is based on work of Levchenko.
3 In both cases, we have modified the method and added significant details.
The synthesis of primary allylamines up to 1983 has been reviewed.
4 Our method involves the ene reaction of various aza analogs of
sulfur or
selenium dioxide (
1 +
2 →
3), followed by [2,3]-sigmatropic rearrangement (
3 →
4). The use of
N-tosyl activating groups, as in our earlier work
5 and that of Sharpless,
6,7,8 has the disadvantage that the
N-sulfonyl group cannot easily be removed under mild conditions, as Sharpless observed in his synthesis of gabaculine.
8 In the present method, the methoxycarbonyl group is used in place of the sulfonyl group. It is easy to remove, but the diimide (
2) is less reactive than the corresponding sulfonyl compound.
9 Nevertheless, it reacts at room temperature with a variety of alkenes, such as
β-methylstyrene,
2-pentene, and
cyclohexene, and more heavily substituted derivatives of these compounds.
10 The urethane group can easily be hydrolyzed or reduced (
lithium aluminum hydride) to give the corresponding allylamines.
10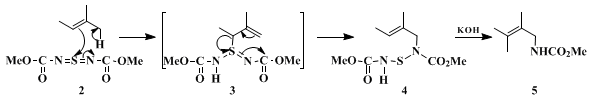
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
calcium chloride (10043-52-4)
acetic acid (64-19-7)
methanol (67-56-1)
ether (60-29-7)
sodium acetate (127-09-3)
chloroform (67-66-3)
Cyclohexene (110-83-8)
sodium chloride (7647-14-5)
nitrogen (7727-37-9)
sulfur (7704-34-9)
pyridine (110-86-1)
chlorine (7782-50-5)
selenium dioxide (7446-08-4)
potassium hydroxide (1310-58-3)
2-Pentene (646-04-8)
β-methylstyrene
magnesium sulfate (7487-88-9)
sulfur dichloride (10545-99-0)
2-methyl-2-butene (513-35-9)
lithium aluminum hydride (16853-85-3)
Carbamic acid, (2-methyl-2-butenyl)-, methyl ester,
Methyl N-(2-methyl-2-butenyl)carbamate (86766-65-6)
methyl carbamate (598-55-0)
bis(methoxycarbonyl)sulfur diimide (16762-82-6)
sulfurdiimide
Methyl N,N-dichlorocarbamate (16487-46-0)
N,N-dichlorocarbamate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved