Org. Synth. 1988, 66, 14
DOI: 10.15227/orgsyn.066.0014
(1-OXO-2-PROPENYL)TRIMETHYLSILANE
[Silane, trimethyl(1-oxo-2-propenyl)-]
Submitted by Rick L. Danheiser, David M. Fink, Kazuo Okano, Yeun-Min Tsai, and Steven W. Szczepanski
1.
Checked by Masahiko Hayashi and Ryoji Noyori.
1. Procedure
Caution! tert-Butylithium is extremely pyrophoric and must not be allowed to come into contact with the atmosphere. This reagent should only be handled by individuals trained in its proper and safe use. It is recommended that transfers be carried out by using a 20-mL or smaller glass syringe filled to no more than 2/3 capacity, or by cannula. For a discussion of procedures for handling air-sensitive reagents, see Aldrich Technical Bulletin AL-134. [Note added August 2009]
A. (1-Hydroxy-2-propenyl)trimethylsilane. A 2-L, three-necked, round-bottomed flask is equipped with a magnetic stirring bar, two pressure-equalizing dropping funnels (250 and 500 mL), and a Claisen adapter fitted with an argon inlet adapter and a rubber septum (Note 1). The flask is charged with 20.0 g (0.344 mol) of allyl alcohol (Note 2), and 400 mL of dry tetrahydrofuran (Note 3), and then cooled below −75°C with a dry ice–acetone bath and maintained at that temperature while 157 mL (0.363 mol) of a 2.31 M solution of n-butyllithium in hexane (Note 4) is added dropwise over 1 hr. After 50 min, a solution of 39.3 g (0.362 mol) of chlorotrimethylsilane (Note 5) in 25 mL of tetrahydrofuran is added dropwise via syringe over 30 min, and the resulting colorless reaction mixture is stirred for 1 hr further, and then treated dropwise over 1.5 hr with 258 mL (0.415 mol) of a 1.61 M solution of tert-butyllithium in pentane (Note 4). After 2 hr of further stirring at −75°C the cold bath is removed, and 100 mL of saturated ammonium chloride solution is added in one portion to the yellow reaction mixture. The resulting solution is stirred for 5 min and then diluted with 50 mL of water and 300 mL of pentane. The organic phase is separated and washed successively with three 100-mL portions of water and two 100-mL portions of saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and concentrated by carefully distilling off the solvents at atmospheric pressure through a 10-cm Vigreux column. The residual pale yellow liquid is transferred to a 100-mL round-bottomed flask, and the remaining volatile impurities are removed by distillation at 15 mm through a 4-cm column packed with glass helices (Note 6), leaving 35.1–39.7 g of (1-hydroxy-2-propenyl)trimethylsilane as a pale-yellow liquid (Note 7) and (Note 8) used in the next step without further purification.
B. (1-Oxo-2-propenyl)trimethylsilane. A 2-L, three-necked, round-bottomed flask is equipped with a mechanical stirrer and two 250-mL pressure-equalizing dropping funnels, one of which is fitted with an argon inlet adapter (Note 1). The flask is charged with 41.71 g (0.329 mol) of oxalyl chloride (Note 9) and 500 mL of dichloromethane (Note 10), and cooled below −75°C with a dry ice–acetone bath and maintained at that temperature while a solution of 55.82 g (0.715 mol) of dimethyl sulfoxide (Note 11) in 60 mL of dichloromethane is added dropwise over 1 hr. After 1 hr, a solution of the crude (1-hydroxy-2-propenyl)trimethylsilane in 100 mL of dichloromethane is added dropwise over 1.25 hr to the colorless reaction mixture, which is stirred at −75°C for 1 hr further, and then treated dropwise over 30 min with 150.38 g (1.486 mol) of triethylamine (Note 12). After 1 hr, the cold bath is removed and the reaction mixture is poured into 200 mL of water. The organic phase is separated and washed successively with five 100-mL portions of 10% hydrochloric acid, three 100-mL portions of water, and two 100-mL portions of saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and concentrated by carefully distilling off the solvents at atmospheric pressure through a 10-cm Vigreux column. The residual yellow oil is transferred to a 250-mL, round-bottomed flask containing 0.050 g of 3-tert-butyl-4-hydroxy-5-methylphenyl sulfide (Note 13) and distilled through a 4-cm column packed with glass helices to afford 27.8–30.0 g (63–68% overall yield based on allyl alcohol) of (1-oxo-2-propenyl)trimethylsilane as a brilliant-yellow oil, bp 47–50°C (30 mm) (Note 14) and (Note 15).
2. Notes
1.
The glass components of the apparatus are dried overnight in a 120°C oven, and then assembled and maintained under an atmosphere of
argon during the course of the reaction.
2.
Allyl alcohol was purchased from Aldrich Chemical Company, Inc. and distilled from
calcium hydride prior to use.
3.
Tetrahydrofuran was distilled from
sodium benzophenone ketyl immediately before use.
4.
n-Butyllithium was purchased from Aldrich Chemical Company, Inc. or Mitsuwa Pure Chemicals,
tert-Butyllithium was obtained from Aldrich Chemical Company, Inc. These were titrated using the method of Watson and Eastham
2 submitters) or Lipton
3 (checkers).
5.
Chlorotrimethylsilane was obtained from Petrarch Systems, Inc., or Shin-etsu Kagaku Co., and distilled from
calcium hydride before use.
6.
The heating bath temperature was not permitted to exceed 70°C during the course of the distillation.
7.
The purity of this material was determined to be 95% by gas-chromatographic analysis (10% OV-101 on 100–120-mesh Chromosorb W, 6 ft × 1/8 in., program: 50°C for 2 min and then 50–250°C at 32°C/min).
8.
The product exhibits the following spectral properties: IR (film) cm
−1: 3420, 2955, 2895, 2820, 1625, 1410, 1245, 1140, 1095, 990, 900, 840;
13C NMR (67.9 MHz, CDCl
3) δ: −4.4, 68.9, 109.4, 139.9;
1H NMR (250 MHz, CDCl
3) δ: 0.05 (s, 9 H), 2.86 (br s, 1 H), 3.88 (m, 1 H), 4.86 (ddd, 1 H,
J = 2, 2, 11), 4.98 (ddd, 1 H,
J = 2, 2, 17), 5.89 (ddd, 1 H,
J = 5.5, 11, 17); HRMS
m/e calcd. for C
6H
14OSi (M
+): 130.0814, Found: 130.0810.
9.
Oxalyl chloride purchased from Aldrich Chemical Company, Inc. was fractionally distilled under
argon before use.
10.
Dichloromethane was distilled from
calcium hydride immediately before use.
11.
Dimethyl sulfoxide was distilled from
calcium hydride immediately before use.
12.
Triethylamine was distilled from
calcium hydride before use.
13.
3-tert-Butyl-4-hydroxy-5-methylphenyl sulfide was purchased from Aldrich Chemical Company, Inc.14.
The purity of this material was determined to be >97% by gas-chromatographic analysis (10% OV-101 on 100–120-mesh Chromosorb W, 6 ft × 1/8 in., program: 50°C for 2 min and then 50–250°C at 32°C/min).
15.
The product exhibits the following spectral properties: IR (film) cm
−1: 2960, 2900, 1635, 1600, 1590, 1415, 1390, 1255, 1185, 985, 960, and 845;
13C NMR (67.9 MHz, CDCl
3) δ: −2.2, 128.5, 141.3, 237.9;
1H NMR (250 MHz, CDCl
3) δ: 0.23 (s, 9 H), 5.94 (dd, 1 H,
J = 1, 11), 6.13 (dd, 1 H,
J = 1, 18), 6.38 (dd, 1 H,
J = 11, 18).
3. Discussion
(1-Oxo-2-propenyl)trimethylsilane has previously been prepared by Reich and co-workers in four steps beginning with
propargyl alcohol.
4 This earlier synthesis proceeded in
45% overall yield and involved as key steps the metalation (at −90°C) and silylation of
1-(1-ethoxyethoxy)-1,2-propadiene, followed by careful hydrolysis of the resulting α-silyl allenyl ether.
The present method
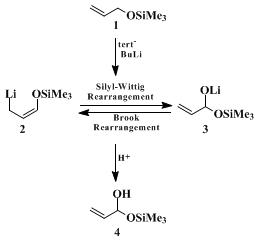
5 offers a more efficient and convenient two-step route to the parent α,β-unsaturated acylsilane derivative. The first step in the procedure involves the conversion of
allyl alcohol to
allyl trimethylsilyl ether, followed by metalation
6 7 8 9 10 11 (in the same flask) with
tert-butyllithium at −75°C. Protonation of the resulting mixture of interconverting lithium derivatives (
2 and
3) with aqueous
ammonium chloride solution furnishes
(1-hydroxy-2-propenyl)trimethylsilane (4), which is smoothly transformed to
(1-oxo-2-propenyl)trimethylsilane by Swern oxidation.
12 The acylsilane is obtained in
63–68% overall yield from
allyl alcohol in this fashion.
α,β-Unsaturated acylsilanes serve as valuable building blocks for the synthesis of a variety of complex organic compounds. These α,β-unsaturated carbonyl derivatives participate in a number of carbon–carbon bond forming processes including organocuprate conjugate additions,
4 TiCl
4-mediated conjugate allylations,
13 Diels–Alder reactions,
4 1,3-dipolar cycloadditions,
14 and the [3+2] annulation method recently developed in our laboratory.
14 The utility of these reactions is enhanced by the fact that the product acylsilanes are subject to a variety of useful further transformations,
15 16 including, for example, Brook reactions,
4,17 18 oxidation to carboxylic acids,
19 20 and fluoride-promoted conversion to ketones and aldehydes.
20,21 22 23 24 The present procedure provides a practical method for the preparation of multigram quantities of the simplest α,β-unsaturated acylsilane.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
sodium benzophenone ketyl
hydrochloric acid (7647-01-0)
ammonium chloride (12125-02-9)
sodium chloride (7647-14-5)
Allyl alcohol (107-18-6)
sodium sulfate (7757-82-6)
Pentane (109-66-0)
dichloromethane (75-09-2)
n-butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
oxalyl chloride (79-37-8)
hexane (110-54-3)
dimethyl sulfoxide (67-68-5)
triethylamine (121-44-8)
argon (7440-37-1)
calcium hydride (7789-78-8)
propargyl alcohol (107-19-7)
CHLOROTRIMETHYLSILANE (75-77-4)
tert-Butyllithium (594-19-4)
(1-Oxo-2-propenyl)trimethylsilane,
Silane, trimethyl(1-oxo-2-propenyl)- (51023-60-0)
(1-Hydroxy-2-propenyl)trimethylsilane (95061-68-0)
1-(1-Ethoxyethoxy)-1,2-propadiene (20524-89-4)
allyl trimethylsilyl ether (18146-00-4)
3-tert-butyl-4-hydroxy-5-methylphenyl sulfide (96-66-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved