Org. Synth. 1990, 69, 188
DOI: 10.15227/orgsyn.069.0188
A GENERAL METHOD FOR THE PREPARATION OF 9-, 10-, AND 11-MEMBERED UNSATURATED MACROLIDES: SYNTHESIS OF 8-PROPIONYL-(E)-5-NONENOLIDE
[2H-Oxecin-2-one, 3,4,5,8,9,10-hexahydro-9-(1-oxopropyl)-, (E)-]
Submitted by Kevin S. Webb, Edward Asirvatham, and Gary H. Posner
1.
Checked by Thais Sielecki and Albert I. Meyers.
1. Procedure
Caution! Benzene has been identified as a carcinogen: OSHA has issued emergency standards on its use. All procedures involving benzene should be carried out in a well-ventilated hood, and gloves should be worn.
A flame-dried (Note 1), 500-mL, three necked, round-bottomed flask equipped with a Teflon-coated magnetic stirring bar, rubber septa, and an argon inlet is charged with 75 mL of anhydrous tetrahydrofuran (Note 2) and 8.0 mL (57.1 mmol) of diisopropylamine (Note 3), then cooled to −10°C via an ice–salt bath. Next 34.8 mL (55.0 mmol) of butyllithium (1.58 M, (Note 4), in hexane) is added dropwise (over 4 min) to the vigorously stirred diisopropylamine solution, and stirred at −10°C for 20 min. Tributyltin hydride (15.2 mL, 55.0 mmol, (Note 5)) is added dropwise over 5 min to the solution, and the reaction mixture is stirred under argon at −10° to 0°C for an additional 30 min. The flask is cooled to −78°C and stirred for 20 min.
A flame-dried (Note 1), 100-mL, round-bottomed flask equipped with a Teflon-coated magnetic stirring bar, a rubber septum, and an argon inlet is charged with 50 mL of anhydrous tetrahydrofuran (Note 2) and 4.85 mL (50.1 mmol) of 2-cyclohexen-1-one (Note 6), and cooled to −78°C. This solution (54.85 mL) is cannulated into the tributyltinlithium solution dropwise over 8 min (Note 7), and the solution is stirred at −78°C under argon for 25 min (Note 8). The same 100-mL, round-bottomed flask is charged with 50 mL of anhydrous tetrahydrofuran (Note 2) and 6.1 mL (59.4 mmol) of ethyl vinyl ketone (Note 9), and cooled to −78°C. This solution (56.1 mL) is cannulated into the 500-mL, round-bottomed flask dropwise over 8 min (Note 10), and the solution is stirred for 1.5 hr (Note 11). The resulting solution is then transferred from a −78°C cold bath to a −23°C cold bath (Note 12) and stirred at this temperature for 30 min.
A flame-dried (Note 1), 100-mL, three-necked, round-bottomed flask equipped with a Teflon-coated magnetic stirring bar, a Teflon stopcock, rubber septa, and an argon inlet is charged with 6 g of paraformaldehyde (Note 13). The flask is heated to 165–170°C and the gaseous formaldehyde is bubbled into the −23°C solution (Note 14). After 15 min all of the paraformaldehyde is pyrolyzed (Note 15), and the solution becomes slightly cloudy and yellow. The 500-mL, three-necked, round-bottomed flask is transferred to a −40°C bath (maintained by a Flexicool cryostat) and allowed to stir under argon for 20 hr. The reaction mixture is quenched at −40°C with 7 mL of saturated ammonium chloride followed by 8 mL of distilled water. It is warmed to room temperature, poured into a 500-mL separatory funnel, and diluted with 75 mL of water; the organic layer is separated, and the aqueous layer is extracted with diethyl ether (2 × 100 mL). The combined organic layers are dried over anhydrous magnesium sulfate, filtered, and concentrated (Note 16) to afford 29.3 g of crude product.
A flame-dried (Note 1), 1000-mL, three-necked, round-bottomed flask equipped with a Teflon-coated magnetic stirring bar, rubber septa, a 24/40 condenser, and an argon inlet is charged with 300 mL of anhydrous benzene (Note 17) and 29.00 g (65.4 mmol) of lead tetraacetate (Note 18). The suspension is heated to 80°C and stirred vigorously under argon. A flame-dried (Note 1), 250-mL, round-bottomed flask equipped with a Teflon-coated magnetic stirring bar, rubber septa, and an argon inlet is charged with 150 mL of anhydrous benzene (Note 17) and 29.3 g of the crude reaction mixture. This solution is cannulated into the lead tetraacetate suspension over 2 min (Note 19), and the suspension is allowed to reflux for 2.5 hr. The reaction flask is cooled to room temperature, quenched with 200 mL of distilled water, poured into a 2-L separatory funnel, and diluted with 1000 mL of diethyl ether (Note 29). The organic layer is washed with saturated sodium bicarbonate solution (3 × 200 mL), aqueous 5% hydrochloric acid (2 × 200 mL), distilled water (200 mL), and brine (200 mL). The organic layer is dried over anhydrous magnesium sulfate and filtered; solvent removal afforded 28.95 g of a crude oil. This crude residue (light-yellow-brown oil) is purified by short-path column chromatography (Note 21) to yield 4.37 g (41.5%) of 8-propionyl-(E)-5-nonenolide (Note 22) and (Note 23).
2. Notes
1.
All glassware and Teflon-coated magnetic stirring bars were flame-dried under vacuum (0.5 mm) for 5 min, then back-filled with
argon. The procedure was repeated a total of 3 times.
2.
Baker reagent-grade tetrahydrofuran (99%, obtained from Aldrich Chemical Company, Inc.) was distilled over
sodium metal spheres/
benzophenone under an inert atmosphere and used immediately.
3.
Diisopropylamine, 99%, was obtained form Aldrich Chemical Company, Inc., and allowed to reflux over
calcium hydride (≥95%, obtained from Aldrich Chemical Company, Inc.) for 24 hr prior to use.
4.
Butyllithium in hexane (1.6 M), obtained from Aldrich Chemical Company, Inc., was titrated (using
2,5-dimethoxybenzyl alcohol as the indicator
2) just prior to use.
5.
Tributyltin hydride, 97%, was obtained form Aldrich Chemical Company, Inc., and must be used quickly to ensure generation of the
tributyltinlithium species. Two minutes after the initial addition of
tributyltin hydride the colorless solution turned light yellow.
6.
2-Cyclohexen-1-one, 97%, was obtained from Aldrich Chemical Company, Inc., and was freshly distilled via short-path distillation.
7.
After the
2-cyclohexenone addition was complete, the 100-mL, round-bottomed flask was washed with
10 mL of anhydrous tetrahydrofuran, and this wash was cannulated into the 500-mL flask
(Note 2).
8.
A small aliquot of the reaction mixture was removed after 15 min and analyzed by analytical TLC. The TLC was developed in
20% ethyl acetate:hexane and showed that the 1,4-conjugate addition had proceeded to completion (
Rf = 0.58); the solution was colorless at this point of the reaction.
9.
Ethyl vinyl ketone, 97%, was obtained from Aldrich Chemical Company, Inc., and used directly.
10.
The reaction mixture turned slightly yellow during addition of
ethyl vinyl ketone.
11.
A small aliquot was removed from the reaction mixture and analyzed by analytical TLC (
20% ethyl acetate:hexane) to ensure that the Michael addition had proceeded,
Rf = 0.44, and
Rf = 0.32 corresponding to the diastereomeric intermediates.
12.
A −23°C bath was obtained from a mixture of dry ice/
carbon tetrachloride. A temperature between −40° and −20°C is necessary.
13.
Paraformaldehyde, 95%, was obtained from Aldrich Chemical Company, Inc., and used directly.
14.
The
argon inlet was equipped with a 16-gauge, 3-in. syringe needle; the transfer cannula was a flex-needle (Z10,091-9) obtained from Aldrich Chemical Company, Inc., and the outlet bubbler was equipped with a 16-gauge, 3-in., syringe needle.
15.
A high-pressure stream of
argon was needed to prevent the gaseous
formaldehyde from polymerizing to
paraformaldehyde in the transfer flex-needle.
16.
The weight of 29.30 g was achieved by attaching the round-bottomed flask to a vacuum (1.5 mm) and heating via a water bath (55°C) for 4 hr.
17.
Benzene (thiophene-free, ≥99%, 900 mL) was obtained from Aldrich Chemical Company, Inc., and washed with concentrated
sulfuric acid (5 × 100 mL), distilled water (100 mL), aqueous
2% sodium hydroxide solution (100 mL), and distilled water (100 mL), and dried over anhydrous
magnesium sulfate. It was then allowed to reflux over
calcium hydride for 24 hr. The checkers found that the
benzene needed to be distilled only from
calcium hydride.
18.
lead tetraacetate was purchased from Aldrich Chemical Company, Inc., and used directly.
19.
The transfer cannula was a 12-in., 16-gauge, double-tipped syringe needle, and the 250-mL flask was washed with an additional
30 mL of anhydrous benzene (Note 17), which was cannulated into the 1000-mL flask.
20.
The 1000-mL, round-bottomed flask was washed with
diethyl ether (3 × 100 mL of the 1000 mL).
21.
Working on one-half of the present scale, the checkers found that by using Amicon Grace Matrex Silica Gel, (60 Å, 20–45 m) and an eluting solvent of
10% diethyl ether:hexane,
3.50 g (
66.0%) of
8-propionyl-(E)-5-nonenolide was obtained. The white solid was further purified by recrystallization from
15 mL of hexane to yield
3.15 g (
60.0%) as a white solid.
22.
The physical properties are as follows: IR (CHCI
3) cm
−1; 2933, 1731, 1349, 1210, 1154, 979, 770, 746;
1H NMR (CDCl
3, 400 MHz) δ: 1.05 (t, 2 H,
J = 7.2), 1.12 (t, 1 H,
J = 7.2), 1.70–3.00 (m, 11 H), 3.76–3.82 (m, 0.67 H), 4.15–4.21 (m, 0.33 H), 5.05–5.70 (m, 3 H). When the proton signals at 1.90–2.90 ppm were irradiated, the two olefinic multiplets collapsed into two doublets (
J = 15.2). An analytical sample (
4.37 g) was recrystallized from
20 mL of hexane (Fisher, certified) to yield
4.05 g (
38.5%) was a white solid: mp
70–71°C. Anal. calcd. for C
12H
18O
3; C, 68.57; H. 8.57. Found: C, 68.47; H, 8.68.
23.
In order to determine whether the product was a mixture of geometrical isomers (e.g., differing by cis or trans geometry at the double bond) or conformers, it was necessary to obtain
1H NMR spectra at various temperatures. The 400 MHz
1H NMR (
d6-DMSO) at 100°C shows that the two triplets (δ 1.05, 1.12) start to collapse to one triplet, the multiplicity of the peaks in the region of δ 5.05–5.70 simplify greatly, and the initial peak ratios in the region of δ 3.70–4.20 change from 1:2 to 1:3. Therefore, the nonenolide is one pure isomer of only
trans double-bond geometry and is able to exist as two stable conformers in solution. The glass capillary gas chromatograph (Hewlett Packard 5890) of
8-propionyl-(E)-5-nonenolide shows only one peak with a retention time of 4.21 min (injector temperature 175°C, detector temperature 225°C).
3. Discussion
The procedure described is a simple, rapid, and convenient method for conversion of
n-sized cycloalkenones into
n + 4 alkenolides. Significant but limited progress has been reported in the recent literature toward the preparation of medium and large-ring lactones via ring-expansion reactions. One of the most notable and useful developments in this area involves conversion of a cycloalkanone into a bicyclic vinylic ether that is oxidatively cleaved to form a ring-enlarged keto lactone.
3,4;
5 Recently, several variations of this ring-enlargement reaction have been reported, including the scission of alkoxy radicals.
6,7,8,9,10,11,12,13,14 In most of these cases, a superfluous functional group (e.g., ketone, iodide), is produced during cleavage of the bicyclic system. Regiospecific conversion of such functional groups into a specific alkene structural unit is usually not possible because of the similar chemical environment α' to the functional group.
15 Because many regiospecifically unsaturated lactones are physiologically active natural products,
16,17,18,19 we have developed methodology to prepare unsaturated macrolides having a carbon–carbon double bond with specific geometry and at a specific position in the macrolide skeleton.
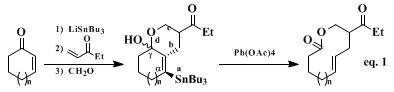
Because of our interest in one-pot, multicomponent annulations,
20,21,22,23,24,25,26 we envisioned a flexible and efficient protocol that would link the four different components via the formation of four new bonds (a–d, Eq. 1) in one reaction vessel. The intermediate γ-hydroxystannanes thus formed in Equation 1 could be oxidatively fragmented
27,28,29 to produce both ring enlargement and regiospecific formation of an alkenyl unit. This four-atom ring-expansion methodology of common side α,β-unsaturated ketones has led to the syntheses of many mono- and disubstituted 9-, 10-, and 11-membered unsaturated macrolides (Table I).
TABLE I
|
|
|
|
n
|
R
|
R'
|
Yield (%)
|
trans:cis
|
|
|
0
|
Et
|
CH3
|
30.5
|
0.57
|
|
1
|
Et
|
CH3
|
47
|
0.7
|
|
1
|
Et
|
CH2=CH
|
39.5
|
2.6
|
|
1
|
Et
|
o-BrC6H4
|
47
|
5.3
|
|
1
|
Et
|
o-IC6H4
|
37
|
2.6
|
|
1
|
Ph
|
o-IC6H4
|
34
|
1.0
|
|
1
|
Et
|
PhCH2
|
43.5
|
1.5
|
|
1
|
Ph
|
o-BrC6H4CH2
|
30
|
1.0
|
|
1
|
Et
|
o-N(phtl)C6H4
|
42
|
5.0
|
|
1
|
Et
|
H
|
41.5
|
—
|
|
1
|
Et
|
2,3-(MeO)2C6H3CH2
|
52
|
1.0
|
|
1
|
Ph
|
2,3-(MeO)2C6H3CH2
|
51
|
1.5
|
|
1
|
Et
|
3-ThienylCH2
|
52
|
0.7
|
|
1
|
Ph
|
3-ThienylCH2
|
52
|
1.8
|
|
1
|
Et
|
3-FurylCH2
|
54
|
0.7
|
|
1
|
Ph
|
3-FurylCH2
|
40
|
7.0
|
|
2
|
Et
|
Cyclopropyl
|
53
|
1.1
|
|
2
|
Et
|
CH3
|
40
|
1.0
|
|
2
|
Et
|
3-(MeO)C6H4
|
41.5
|
1.5
|
|
2
|
Ph
|
o-IC6H4CH2
|
38
|
1.9
|
|
Based on the data in Table I and on our published results,
20,21,22,23,24,25,26 it is clear that five-, six-, and seven-membered cycloalkenones undergo this four-atom ring enlargement reaction to produce medium ring, unsaturated lactones in overall yields of 30–54%. Permutations on this methodology include using either
ethyl vinyl ketone or
phenyl vinyl ketone as the third component, and either substituted acetaldehydes or substituted benzaldehydes as the last component. The geometrical assignment of the new carbon–carbon double bond was made from interpreting the 400 MHz
1H NMR decoupled spectra in which each olefinic proton collapsed into a baseline-resolved doublet with coupling constants of
J = 15–16. The proton decoupling experiments conducted to determine the relative stereochemistry of the vicinal substituents in the disubstituted macrolides were inconclusive; often the magnitude of the coupling constants were similar or not discernible from the spectra. Therefore, the relative stereochemistry of the vicinal substituents was established by examining the
1H NMR spectra of the intermediate γ-hydroxystannanes (usually only two were isolated). The
trans-hemiketals showed typical coupling constants of
J = 8–13, while the
cis-hemiketals showed coupling constants of
J = 2–4. Separate
lead tetraacetate oxidative fragmentation of these γ-hydroxystannanes produced two different ring-enlarged lactones, both with specific
trans double-bond geometry and differing only in the relative stereochemistry of vicinal substituents.
This homologous Baeyer–Villiger-type oxidative ring expansion represents a conceptually new protocol illustrating the substantial value of one-pot, four-component annulations as a flexible and simple new synthetic method.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
brine
sulfuric acid (7664-93-9)
hydrochloric acid (7647-01-0)
Benzene (71-43-2)
ethyl acetate (141-78-6)
diethyl ether (60-29-7)
ammonium chloride (12125-02-9)
sodium hydroxide (1310-73-2)
formaldehyde (50-00-0)
sodium bicarbonate (144-55-8)
carbon tetrachloride (56-23-5)
Benzophenone (119-61-9)
sodium (13966-32-0)
magnesium sulfate (7487-88-9)
butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
hexane (110-54-3)
argon (7440-37-1)
tributyltin hydride (688-73-3)
calcium hydride (7789-78-8)
2-Cyclohexenone,
2-cyclohexen-1-one (930-68-7)
cyclopropyl (2417-82-5)
diisopropylamine (108-18-9)
2,5-dimethoxybenzyl alcohol (33524-31-1)
Tributyltinlithium
ethyl vinyl ketone (1629-58-9)
phenyl vinyl ketone (768-03-6)
8-Propionyl-(E)-5-nonenolide
2H-Oxecin-2-one, 3,4,5,8,9,10-hexahydro-9-(1-oxopropyl)-, (E)- (114633-68-0)
lead tetraacetate (546-67-8)
paraformaldehyde (30525-89-4)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved