Org. Synth. 1997, 74, 229
DOI: 10.15227/orgsyn.074.0229
RHODIUM-CATALYZED HETEROCYCLOADDITION OF A DIAZOMALONATE AND A NITRILE:
4-CARBOMETHOXY-5-METHOXY-2-PHENYL-1,3-OXAZOLE
[4-Oxazolecarboxylic acid, 5-methoxy-2-phenyl-, methyl ester]
Submitted by Joshua S. Tullis and Paul Helquist
1.
Checked by Anne K. Courtney and Stephen F. Martin.
1. Procedure
CAUTION! This experiment should be conducted behind an auxiliary safety shield in a closed fume hood because of the explosive nature of diazo and azide compounds.
A. Dimethyl diazomalonate. Into a 500-mL, round-bottomed flask is placed a large magnetic stirring bar (Note 1), 4-acetamidobenzenesulfonyl azide (72.3 mmol, 17.36 g, (Note 2)), acetonitrile (250 mL, (Note 3)), and triethylamine (76.5 mmol, 7.74 g, (Note 4)). The reaction mixture is cooled to 0°C, and dimethyl malonate (72.0 mmol, 9.51 g, (Note 4)) is slowly added over 3–5 min by a syringe. The reaction mixture is kept at 0°C for 10 min then stirred at room temperature for 17 hr. A thick white precipitate that forms during the reaction is removed by filtration through a Büchner filter. The filter cake is washed with a 1:1 mixture of petroleum ether and ethyl ether (200 mL, (Note 5)). The resulting filtrates are concentrated by rotary evaporation under reduced pressure giving a yellow oil with some visible precipitate of sulfonamide. Repeated trituration and filtration of this oil with 1:1 petroleum ether and ethyl ether (6 × 50 mL, (Note 5)) to remove the solid 4-acetamidobenzenesulfonamide, followed by concentration of all filtrates by rotary evaporation under reduced pressure, gives 10.92 g of the crude diazo product as a yellow oil (96%). The crude product is purified by distillation to provide 8.26 g (73%) of dimethyl diazomalonate as a bright yellow oil (Note 6).
B. 4-Carbomethoxy-5-methoxy-2-phenyl-1,3-oxazole. In an oven-dried, 100-mL, round-bottomed flask is placed a small magnetic stirring bar (Note 7), rhodium(II) acetate dimer, Rh2(OAc)4, (0.230 mmol, 0.102 g, (Note 4)), benzonitrile (19.5 mmol, 2.011 g, (Note 8)), and chloroform (CHCl3, 5 mL, (Note 9)). The flask is placed in an oil bath and fitted with a 30-cm reflux condenser. On top of the condenser is placed a three-way stopcock having horizontal and vertical tubulations. A source of nitrogen (N2) is connected to the system via the horizontal tubulation of the stopcock, and a septum is placed over the vertical tubulation. The entire system is flushed several times with N2, and the solution is heated to reflux (68°C) during which time it becomes deep purple.
Into a dry, 50-mL, conical vial is placed dimethyl diazomalonate (29.4 mmol, 4.65 g) and 25 mL of chloroform (Note 9). The solution is taken up into a syringe (Note 10) fitted with a 30-cm needle. The needle is inserted about half way into the septum on the stopcock so that the needle tip extends down into the condenser. The syringe is placed on a syringe pump (Note 11) which is set to add the solution at a rate of 1.17 mL/hr over a period of about 25 hr. After the addition is complete, the syringe is again filled with 2 mL of CHCl3, which is added to the system via the septum. The needle is removed, and the stopcock plug is rotated so that the septum is closed off from the system while the N2 inlet remains open. The solution is allowed to continue stirring at reflux for another 20 hr. It is then concentrated by rotary evaporation under reduced pressure to yield a brownish-purple oil containing crude product. The crude oxazole is purified using flash column chromatography (silica gel, 40% ethyl acetate in hexane, (Note 12), (Note 13), (Note 14)) to yield 2.73–2.96 g (60–65%) of 4-carbomethoxy-5-methoxy-2-phenyl-1,3-oxazole as white needles (Note 15).
2. Notes
1.
An
egg-shaped stirring bar ca. 2.5 cm in length is recommended for stirring the suspension that is produced during the reaction.
2.
4-Acetamidobenzenesulfonyl azide is obtained from Aldrich Chemical Company, Inc., and used without further purification. In addition,
4-acetamidobenzenesulfonyl azide can be synthesized from the precursor,
4-acetamidobenzenesulfonyl chloride.
23.
The checkers used standard
reagent grade acetonitrile from EM Science.
4.
Triethylamine, dimethyl malonate, and rhodium(II) acetate dimer were purchased from Aldrich Chemical Company, Inc., and used without further purification.
5.
Standard
reagent grade ether and petroleum ether are used during workups.
6.
Crude
dimethyl diazomalonate is distilled under reduced pressure using a
vacuum pump and a short-path distillation apparatus equipped with a
Vigreux fractionating column measuring 8–10 cm. The apparatus is wrapped in glass wool and
aluminum foil to minimize heat loss. Pure
dimethyl diazomalonate3 4 5 is a bright yellow oil: bp
60°C, ca. 0.65 mm (lit.
5 bp
45°C, 0.2 mm);
1H NMR (500 MHz, CDCl
3) δ: 3.79 (s, 6 H, COOCH
3; lit.
5 δ 3.76);
13C NMR (125 MHz, CDCl
3) δ: 52.4, 65.6, 161.3; IR (neat) cm
−1: 3006 (m), 2958 (s), 2849 (w), 2138 (s), 1760 (s), 1738 (s), and 1696 (s); (lit.
3,4,5 IR).
7.
The specific size of the stirring bar is not important, but the stirring rate should be adjusted so that the
Rh2(OAc)4 catalyst remains suspended in solution. It should not be resting on the bottom of the round-bottomed flask, nor should it be splashed onto the sides of the flask.
8.
Benzonitrile is available from Aldrich Chemical Company, Inc. and is distilled at reduced pressure (0.5 mm, 75°C) using a short-path distillation apparatus.
9.
Spectroscopic grade chloroform is obtained from J. T. Baker, Inc., (99+% purity) and used without further purification.
10.
The syringe used is a 50-mL, gas-tight syringe (Model #1050) available from Hamilton Company. It is very important that a gas-tight syringe be used during the addition because of the tendency of
chloroform to leak with loss of starting material from other types of syringes.
11.
The syringe pump used is available from Sage Instruments, Model Number 341B.
NOTE: The slow addition of the
dimethyl diazomalonate is of key importance to the success of the reaction. When scaling this reaction up or down, the rate of addition should be altered so that the addition time remains constant at ca. 25 hr. For example, in the reaction described here, there is ca. 30 mL of solution containing
dimethyl diazomalonate, and it is added over 25 hr at a rate of 1.17 mL/hr. If the reaction were at 20% scale, the rate of addition would be 0.23 mL/hr.
12.
On one occasion, crude product was a solid and was dissolved in a minimum amount of eluent and added to the top of the silica column.
13.
To purify the product on this scale, a
35-mm wide column with a
500-mL glass bulb on top and a glass frit at the base is used. It is packed with
25–30 cm silica gel 60 (230–400 mesh, available from EM Science) and eluted with solvent to remove any air bubbles. The purification is monitored by TLC (silica gel,
40% ethyl acetate in hexanes; R
f = 0.35).
14.
Usually, the purification leads to several fractions containing a mixture of the product and the starting material,
dimethyl diazomalonate, (R
f = 0.43). In this event, an alternative to performing another column chromatography has been developed based on trituration according to the following procedure. All fractions that contain the mixture are combined in a round-bottomed flask, and the solvent is removed by rotary evaporation under reduced pressure. The mixture should appear to be a yellow oil mixed with the crystalline oxazole. With an
ice bath, the mixture is cooled along with a separate flask containing 10–15 mL ether. Using a
pipette,
2–3 mL of the cold ether is transferred to the mixture. The mixture is swirled, and the solvent is removed with a pipette. This procedure is repeated several times until only white crystals remain. It is important to keep both flasks cool throughout the procedure so that loss of the product is minimized.
15.
The product,
4-carbomethoxy-5-methoxy-2-phenyl-1,3-oxazole, is obtained as colorless needles: mp
92–95°C (lit.
6 mp
98–99°C);
1H NMR (500 MHz, CDCl
3) δ: 3.86 (s, 3 H, COOCH
3), 4.20 (s, 3 H, C=COCH
3), 7.36–7.39 (m, 3 H, Ar-H), 7.89–7.93 (m, 2 H, Ar-H);
13C NMR
6 (125 MHz, CDCl
3) δ: 51.7 (COOCH
3), 59.7 (C=COCH
3), 107.4 (C-4), 125.8 (C-3', C-5'), 126.2 (C-1'), 128.6 (C-2', C-6'), 130.3 (C-4'), 150.8 (C-5), 161.7, 161.8 (COOCH
3, C-2); IR (CDCl
3) cm
−1: 1713 (COOCH
3), 1619 (C=COCH
3); mass spectrum
6 (Cl, argon) m/e (rel intensity) 233 (M
+, 27), 173 (11), 146 (15), 105 (100), 77 (24). Anal Calcd for C
12H
11NO
4: C, 61.80; H, 4.75. Found: C, 61.53; H, 4.75.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Oxazoles are well-investigated compounds. The occurrence, uses, and synthesis of oxazole derivatives have been the subjects of extensive reviews.
7 8 9 10 The heterocyclic oxazole unit is seen with various substitution patterns in a large number of naturally occurring compounds. Furthermore, oxazoles serve as synthetic intermediates leading to many other systems.
7,8,9,10,11 12,13 14 15 16 17 In this context, oxazoles have seen, for example, numerous applications as "2-azadiene" components in 4+2 cycloadditions with several types of dienophiles. Further transformations of the products then lead to a number of other nitrogen- or oxygen-containing heterocyclic products.
11,12Of the many specific types of oxazoles, those bearing a 4-carboxy-derived group are of considerable importance. Among these compounds are several natural products, including many which contain this moiety, or the analogous thiazole group. Two examples, from the type A streptogramin family, include virginiamycin M
1 and madumycin I.
18 19 20,21 22 23 24 25 26Because of the many roles that these compounds play in natural products and synthetic organic chemistry, many methods have been developed for the preparation of 4-carboxy derivatives of oxazoles.
7,8,9,10,27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 A well-established, classical method is the Cornforth oxazole synthesis (Scheme 1). This method has been shown to be widely applicable, although it employs a complex, multi-step pathway.
47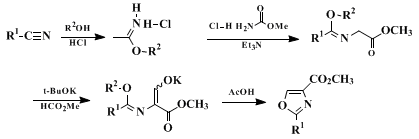
In consideration of conceivable strategies for the more direct construction of these derivatives, nitriles can be regarded as simple starting materials with which the 3+2 cycloaddition of acylcarbenes would, in a formal sense, provide the desired oxazoles. Oxazoles, in fact, have previously been obtained by the reaction of diazocarbonyl compounds with nitriles through the use of
boron trifluoride etherate as a Lewis acid promoter. Other methods for attaining oxazoles involve thermal, photochemical, or metal-catalyzed conditions.
48 49 50 51 52 53 54 55 56 Several recent studies have indicated that many types of rhodium-catalyzed reactions of diazocarbonyl compounds proceed via formation of electrophilic
rhodium carbene complexes as key intermediates rather than free carbenes or other types of reactive intermediates.
57 58 59 60 If this postulate holds for the reactions described here, then the mechanism outlined in Scheme 2 may be proposed, in which the carbene complex
3 and the adduct
4 are formed as intermediates.
61 62 63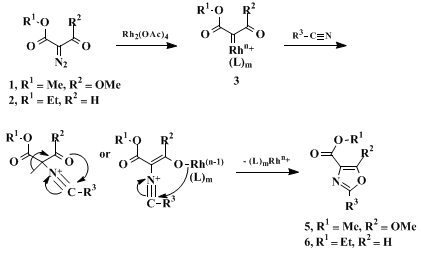
The particular reaction described in Scheme 2 using
dimethyl diazomalonate produces oxazoles
5 that bear a methoxy group at C-5. If desired, this substituent may be removed in some cases by reductive cleavage using
LiB(Et)3H to give the 5-unsubstituted oxazoles
6.
3,4,5,64 Alternatively, the 5-unsubstituted derivatives
6 may be obtained directly through the use of
diazo formylacetate (2) in place of
dimethyl diazomalonate (1).
3,4,5,64 Some additional, representative examples of the use of
1 and
2 are shown below in the Table.
TABLE
ADDITIONAL EXAMPLES OF OXAZOLE FORMATION USING DIMETHYL DIAZOMALONATE 1 AND DIAZO FORMYLACETATE 2a
|
|
Nitrile
|
Diazo Compound
|
Product
|
Yield
|
|
|
3-ClC6H4CN
|
1
|
|
96
|
|
4-CH3C6H4CN
|
1
|
|
93
|
|
Ph-CN
|
2
|
|
45
|
|
BrCH2CN
|
2
|
|
65
|
|
aResults taken from references 6 and 64.
|
In conclusion, the rhodium-catalyzed reaction of diazo dicarbonyl compounds with nitriles provides direct access to useful classes of functionalized oxazoles. This direct, one-step pathway provides a significant improvement over past preparations.
Acknowledgment. We wish to express our appreciation to the National Institutes of Health and the National Science Foundation for providing generous financial support for this work.
This preparation is referenced from:
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
petroleum ether
Rh2(OAc)4
3-ClC6H4CN
4-CH3C6H4CN
Ph-CN
ethyl acetate (141-78-6)
ether,
ethyl ether (60-29-7)
acetonitrile (75-05-8)
benzonitrile (100-47-0)
4-acetamidobenzenesulfonyl chloride (121-60-8)
chloroform,
CHCl3 (67-66-3)
nitrogen (7727-37-9)
aluminum (7429-90-5)
hexane (110-54-3)
triethylamine (121-44-8)
boron trifluoride etherate (109-63-7)
dimethyl malonate (108-59-8)
oxazole,
1,3-oxazole (288-42-6)
(5503-41-3)
4-Carbomethoxy-5-methoxy-2-phenyl-1,3-oxazole,
4-Oxazolecarboxylic acid, 5-methoxy-2-phenyl-, methyl ester (53872-19-8)
Dimethyl diazomalonate (6773-29-1)
4-acetamidobenzenesulfonyl azide (2158-14-7)
4-acetamidobenzenesulfonamide (121-61-9)
rhodium carbene
diazo formylacetate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved