Org. Synth. 1996, 73, 184
DOI: 10.15227/orgsyn.073.0184
DIETHYL (2S,3R)-2-(N-tert-BUTOXYCARBONYL)AMINO- 3-HYDROXYSUCCINATE
[D-Aspartic acid, N-[(1,1-dimethylethoxy)carbonyl]-3-hydroxy-, diethyl ester, threo-]
Submitted by Seiki Saito, Kanji Komada, and Toshio Moriwake
1.
Checked by Martin Fox and Larry E. Overman.
1. Procedure
Caution! Parts A and C of this procedure should be carried out in a well-ventilated hood since toxic hydrogen bromide and azidotrimethylsilane are handled and toxic hydrogen azide is liberated during the course of the reaction.
A. Diethyl (2S,3S)-2-bromo-3-hydroxysuccinate (2). In an oven-dried, 300-mL, round-bottomed flask containing a Teflon-coated stirring bar is placed 26.0 g (0.126 mol) of diethyl L-tartrate (Note 1). A pressure-equalizing dropping funnel containing 100 mL (0.504 mol) of 30% hydrobromic acid (HBr) in acetic acid (Note 2) is mounted on the flask and the top of the funnel is fitted with a nitrogen inlet vented through an oil bubbler. The system is placed under a nitrogen atmosphere, magnetic stirring is initiated, and contents of the flask are cooled in an ice-water bath. The contents of the dropping funnel are added to the cooled tartrate during 30 min and the yellow mixture is stirred for an additional 15 min after completion of the addition. The cooling bath is removed and the reaction mixture is allowed to reach 25°C and stir for an additional 10 hr or until disappearance of the tartrate as judged by TLC analysis (Note 3). The light brown reaction mixture is poured into 500 g of ice, and the resulting mixture is transferred to a 1-L separatory funnel and extracted four times with 80 mL of ether. The combined ether extracts are washed successively three times with 60 mL of water, and then 100 mL of brine, dried over magnesium sulfate, filtered, and concentrated under reduced pressure using a rotary evaporator to give a pale yellow oil.
A
single-necked, 300-mL, round-bottomed flask, equipped with a Teflon-coated stirring bar and an
efficient reflux condenser bearing a
drying tube packed with blue indicator silica gel beads, is charged with the yellow oil obtained above and
140 mL of ethanol (Note 4) to which is cautiously added
4 mL of acetyl chloride with stirring
(Note 5). The mixture is heated under gentle reflux for 7 hr, then cooled to room temperature, the condenser is removed, and the mixture is concentrated under reduced pressure at 50–60°C using a rotary evaporator to give a yellow oil. The crude product is transferred to a silica gel column
(Note 6) with the aid of a small amount of
hexane-ethyl acetate (4:1) and eluted with the same mixed solvent in 60-mL fractions. Fractions 5 to 15
(Note 7) are combined and concentrated on a rotary evaporator to give a colorless oil, which, upon vacuum distillation, affords
24.36–25.73 g (
72–76%) of 99% pure (GLC)
diethyl (2S,3S)-2-bromo-3-hydroxysuccinate, bp
93–94°C (0.2 mm) [lit.
2bp
123–125°C (0.6 mm)],
[α]25.6D −16.8° (EtOH,
c 5.70),
[α]27D −35.72° (neat) [lit.
2[α]21D −28.9° (neat)]
(Note 8),
(Note 9).
B.
Diethyl (2R,3R)-2,3-epoxysuccinate (
3). An oven-dried, 300-mL, round-bottomed flask, equipped with a Teflon-coated stirring bar and a
rubber septum through which is inserted a
large bore, needle-tipped nitrogen line vented through an oil bubbler, is charged with
29.7 g (0.110 mol) of diethyl (2S,3S)-2-bromo-3-hydroxy succinate and
80 mL of dry ethanol (Note 10). In a separate,
oven-dried, 250-mL, round-bottomed flask, capped with a rubber septum and vented though a bubbler as described above, a solution of
sodium ethoxide in ethanol is prepared from
3.05 g of sodium (0.132 mol) and
120 mL of dry ethanol (Note 10). (
Caution: This operation should be conducted in a well-ventilated hood. Sodium is a highly reactive metal, avoid exposure to moisture. Hydrogen gas, which is highly flammable and forms explosive mixtures with air, is rapidly evolved. External cooling may be necessary.) The two flasks are placed under a
nitrogen atmosphere, and connected by means of a long double needle-tipped cannula. Magnetic stirring is initiated, and the ethanolic solution of the succinate
2 is cooled in an ice-water bath for 15 min followed by dropwise addition of the solution of
sodium ethoxide in
ethanol which is transferred via the cannula under a very slight positive pressure of
nitrogen (Note 11). Complete transfer of the base should require 2 hr
(Note 12). The reaction mixture is stirred for an additional 20 min at 0–10°C (bath temperature) and then quenched by the addition of
1.43 mL (0.025 mol) of acetic acid. The reaction mixture is diluted with 900 mL of water, transferred to a
2-L separatory funnel, and extracted four times with
100 mL of dichloromethane. The combined organic phases are washed with
100 mL of brine, dried over
sodium sulfate, filtered, and concentrated under reduced pressure using a rotary evaporator to give a colorless oil. Vacuum distillation of this crude product affords
17.7–18.7 g (
85–90%) of
diethyl (2R,3R)-2,3-epoxysuccinate (3), bp
97–98°C (0.9 mm) [lit.
2bp
100–104°C (4 mm)],
[α]25D −110° (EtOH,
c 5.09),
[α]28D −115° (neat) [lit.
2 [α]21.5D −88.47° (
ether,
c 1.030)] which is homogeneous by GLC analysis
(Note 9), and
(Note 13),
(Note 14),
(Note 15).
C. Diethyl (2S,3R)-2-azido-3-hydroxysuccinate (4). An oven-dried, 200-mL, round-bottomed flask, containing a Teflon-coated stirring bar, is capped with a septum while hot, and purged with nitrogen until cool via insertion through the septum of a needle-tipped nitrogen line (vented through an oil bubbler) and an open needle. The open needle is removed and the flask is charged with a solution of 3.45 g (0.0282 mol) of 4-(dimethylamino)pyridine (DMAP) in 17 mL of N,N-dimethylformamide (DMF) (Note 16) and (Note 17) and 1.93 mL (0.0329 mol) of dry ethanol (Note 11) through the septum via syringe. Stirring is initiated and the mixture is cooled in an ice-water bath. To this cold solution is added 17.1 mL (0.122 mol) of azidotrimethylsilane (Note 18) dropwise at 0°C by means of a syringe (Note 19). The heterogeneous mixture is stirred for 15 min at room temperature (25°C) and then a solution of 17.7 g (0.0940 mol) of diethyl (2R,3R)-2,3-epoxysuccinate (3) in 50 mL of chloroform (Note 20), prepared in a septum-capped, oven-dried, 100-mL, round-bottomed flask, is rapidly transferred via cannula with the aid of a positive nitrogen pressure as above (Part B). The mixture is stirred at 25°C until epoxide 3 can no longer be detected upon TLC analysis of the reaction mixture (~40 hr). The mixture is then diluted with 300 mL of water, transferred to a 1-L separatory funnel, and the organic phase separated. The aqueous phase is extracted four times with 60 mL of a hexane-ether mixture (1:1 v/v). The combined organic phases are added to 20 mL of a magnetically stirred 2.3 N solution of hydrogen chloride in ethanol (Note 21), followed by dilution of the resulting mixture with an additional 50 mL of ethanol and continued stirring at room temperature for 30 min (Note 22). After transfer to a separatory funnel, the solution is washed successively with four 50-mL portions of water, 50 mL of saturated aqueous sodium bicarbonate solution, and 50 mL of saturated aqueous sodium chloride solution, dried over sodium sulfate, and concentrated under reduced pressure using a rotary evaporator followed by exposure to high vacuum affording 18.6 g (86%) of 96–98% pure diethyl (2S,3R)-2-azido-3-hydroxysuccinate (4) as a pale yellow oil, [α]20D +31.8° (EtOH, c 18.1), as judged by NMR analysis (Note 23) and (Note 24). This material contains 2–4% of the inseparable (2R,3R)-diastereomer (Note 24).
D.
Diethyl (2S,3R)-2-(N-tert-butoxycarbonyl)amino-3-hydroxysuccinate (
5). A
500-mL, single-necked, round-bottomed flask, equipped with a Teflon-coated stirring bar, is charged with a suspension of
0.91 g of 10% palladium on carbon catalyst
(Note 25) in
100 mL of ethyl acetate (Note 26). The flask is connected to a normal pressure hydrogenation apparatus
(Note 27) and the catalyst is saturated with
hydrogen. After removal of the
hydrogen, a solution of
18.2 g (0.0785 mol) of 4 and
20.6 g (0.0942 mol) di-tert-butyl dicarbonate (Note 28) in 80 mL of ethyl acetate (Note 26) is added to the suspension of catalyst, a
hydrogen atmosphere reestablished, and the suspension is stirred at room temperature under a slight positive pressure of
hydrogen for 4–6 hr
(Note 29). The suspension is filtered through a Celite pad, and the pad is rinsed with several portions of
ethyl acetate. The combined
ethyl acetate solutions are concentrated on a rotary evaporator and finally under high vacuum to give a pale yellow oil that is initially purified by means of a column packed with silica gel (100 g) using
hexane-
ethyl acetate (6:1) as eluent
(Note 30). Fractions containing the product are combined and concentrated on a rotary evaporator to give
23.3 g of crude
5 as a colorless oil. The oily crude
5 is dissolved in
70 mL of hexane-ether (3:1), and the solution is cooled to −30°C, seeded, and kept overnight at that temperature (freezer) to allow crystallization
(Note 31). The mother liquor is siphoned out while the mixture is kept at −30°C (
dry ice-acetone bath). The crystals are washed with several portions of
hexane-
ether (3:1) at −30°C, then dried under high vacuum to provide
12.2–12.7 g of diastereomerically and enantiomerically pure
diethyl (2S,3R)-2-(N-tert-butoxycarbonyl)amino-3-hydroxysuccinate (5) as colorless prisms, mp
33–34°C; [α]24D +13.1° (EtOH,
c 6.32);
[α]24D +28.7° (CHCl
3,
c 6.05)
(Note 32). The combined mother liquor and the
hexane–
ether (3:1) washings are concentrated on a rotary evaporator to give a colorless oil, which upon crystallization as above provides an additional
2.7–3.8 g of product
5 (Note 33). The combined yield of crystalline
5 is
15.9–16.5 g (
66–73%).
2. Notes
1.
Diethyl L-tartrate was usually purchased from Aldrich Chemical Co., Ltd. The diester was also prepared from
L-tartaric acid by the procedure of Kocienski
3 (
triethyl orthoformate–
ethanol–
acetyl chloride) or Seebach
4 [ethanol–acidic ion-exchange resin (Lewatit 3333)].
2.
A solution of
HBr (30%) in acetic acid (d = 1.31 g/mL) was purchased from Kishida Chemical Co., Ltd. (Japan). When 3 equiv of
HBr rather than 4 equiv was employed, the yield of
2 dropped by more than
40%.
3.
Using Merck precoated silica gel plates (0.25-mm thickness) with elution by
hexane-
EtOAc (2:1), after this time period, the starting tartrate ester (R
f = 0.14) was absent. Three spots with R
f values of 0.53 (the largest), 0.36 (half of the largest), and 0.21 (trace) were observed upon visualization. See discussion for the structure of these products.
4.
Commercial reagent grade ethanol was used as received.
5.
Acetyl chloride was purchased from Kishida Chemical Co. Ltd. (Japan), and used as received.
6.
A
column (350 mm × 50 mm) packed with Merck silica gel 60 (230–400 mesh ASTM) was used. Elution was facilitated by the action of an
air pump for tropical fish tanks available from supermarkets. After use, the column was washed successively with
methanol (500 mL),
ethyl acetate (400 mL), and
hexane (400 mL). Such a washing operation reactivates the column enough for reuse to purify the epoxide (
3), if necessary (see
(Note 15)). Thus the submitters can employ the same column repeatedly for the purification of
2 and
3 (over 10 times). The checkers recommend purification by flash chromatography using a
350-mm × 50-mm column.
7.
Fractions 17 to 27, 25–27 eluted with
hexane-
ethyl acetate (1:2), afforded recovered
diethyl L-tartrate (3.1 g).
8.
The spectral data for
2 are as follows:
1H NMR (500 MHz, CDCl
3) δ: 1.31 (t, 6 H, J = 7.1), 3.42 (d, 1 H, J = 7.5), 4.21–4.35 (m, 4 H), 4.66 (dd, 1 H, J = 7.5, 4.1), 4.71 (d, 1 H, J = 4.1);
13C NMR (50 MHz, CDCl
3) δ: 13.7, 13.8, 47.4, 62.3, 62.6, 72.3, 166.5, 170.1; IR (film) cm
−1: 3480, 2909, 2873, 1739, 1371, 1302, 1286, 1220, 1161, 1113, 1024.
9.
A Simadzu GC-8A gas chromatograph equipped with a flame ionization detector and ULBON HR-101
capillary column (23 m × 0.25 mm) was used.
10.
Ethanol was freshly distilled from
magnesium ethoxide.
11.
If it proves difficult to control the rate of addition by adjusting the
nitrogen pressure, a static positive pressure of
nitrogen can be employed and the flask containing the
sodium ethoxide solution can be moved up or down slightly to regulate the rate of addition.
12.
More rapid addition of the base results in a lower yield of
3.
13.
The spectral data for
3 are as follows:
1H NMR (300 MHz, CDCl3) δ: 1.31 (t, 6 H, J = 7.1), 3.66 (s, 2 H), 4.21–4.32 (m, 4 H);
13C NMR (50 MHz, CDCl
3) δ: 13.9, 51.9, 62.1, 166.6; IR (film) cm
−1: 2988, 1751, 1372, 1330, 1200, 1029.
14.
Epoxide
3 should be free from
2 because
2, if subjected to the next step (
3 →
4), leads to the (2R,3R)-diastereoisomer via an S
N2 displacement process. If
3 is contaminated with
2, the distillate can be purified using the same column as above
(Note 6), employing
hexane–
ether (5:1) as eluent and 60-mL fractions. In the checkers' hands a trace of
2 was always present. Therefore, the checkers recommend that the crude product be purified by flash chromatography (as in step A) to remove residual
2 prior to distillation.
15.
The literature values
2 for the antipode (95% pure by GLC analysis) are as follows: bp 98–99°C (3 mm) and [α]
23D +105.49° (ether,
c 1.413).
16.
DMAP was purchased from Nacalai Tesque, Inc. (Japan) and used as received.
17.
N,N-Dimethylformamide was distilled from
calcium hydride.
18.
Azidotrimethylsilane was purchased from Aldrich Chemical Co., Ltd. (95% pure) and used as received.
19.
The reaction of
azidotrimethylsilane and
ethanol, which gives
hydrogen azide and
ethoxytrimethylsilane, is exothermic and the
hydrogen azide immediately forms a salt with
4-(dimethylamino)pyridine, a part of which cannot be dissolved in
N,N-dimethylformamide and precipitates.
20.
Commercial, reagent grade chloroform was used as received.
21.
The solution was prepared by carefully mixing
3.28 mL acetyl chloride with sufficient dry
ethanol to reach a final volume of 20 mL.
22.
This operation transforms any
diethyl (2S,3R)-2-azido-3-(trimethylsiloxy)succinate present to the desired
diethyl (2S,3R)-2-azido-3-hydroxysuccinate.
23.
The optical rotation of
4 is highly dependent both on solvent and sample concentration. For example, [α]
D values observed in
chloroform vary as follows: +1.43° (
c 3.25) , +3.69° (
c 6.61), and +5.68° (
c 16.7).
24.
The spectral data for
4 are as follows:
1H NMR (500 MHz, CDCl
3) δ: 1.30 (t, 3 H, J = 7.1), 1.31 (t, 3 H, J = 7.1), 3.38 (d, 1 H, J = 5.4), 4.20–4.34 (m, 5 H), 4.63 (dd, 1 H, J = 5.4, 2.7);
13C NMR (50 MHz, CDCl
3) δ: 13.90, 13.94, 62.3, 62.6, 64.3, 72.0, 166.9, 170.7; IR (film) cm
−1: 3480, 2989, 2943, 2910, 2877, 2120, 1751, 1724, 1209, 1114, 1028. The signal corresponding to C
3 proton of the (2R,3R) diastereoisomer appears at δ 4.73 (dd, J = 5.6, 2.4).
25.
10% Palladium on carbon catalyst was purchased from Ishizu Seiyaku Co., Ltd. (Japan) and employed as received.
26.
Commercial reagent-grade ethyl acetate was used as received.
Ethyl acetate is the best choice of solvent with regard to reaction time. Alcohols such as
methanol or
ethanol should be avoided because
di-tert-butyl dicarbonate is decomposed by these solvents.
27.
An MRK catalytic hydrogenation apparatus (Mitamura Riken Co., Japan) was used.
28.
Di-tert-butyl dicarbonate (97% pure) was purchased from Wako Pure Chemical Industries, Ltd. (Japan) and used as received.
29.
The time required for completion of the reaction depends on the activity of the
palladium catalyst employed.
30.
This solvent system is convenient to separate less polar contaminants such as tert-butyl acetate or unchanged
di-tert-butyl dicarbonate.
31.
The seeds required for crystallization are available by cooling a small portion (
1 mL) of the hexane–ether solution to −78°C and scratching the highly viscous gum that separates with a
glass rod. The rate of crystallization is very slow and additional
ether is sometimes required when an oil phase appears on cooling instead of development of crystals.
32.
When commercial
chloroform (stabilized with
0.3–1% of ethanol) was used as received for optical rotation measurements, the submitters obtained
[α]24D +19.3° (
c 6.05). However,
chloroform passed through a silica gel column prior to the measurement gave a much higher value as indicated. The optical rotation of
5 observed with commercial
ethanol was (
[α]24D +19.3° (EtOH,
c 6.32)), while that determined using absolute EtOH [distilled from Mg(OEt)
2] led to a much lower value
[α]24D +7.19° (EtOH,
c 6.32). The spectral properties for pure
5 are as follows:
1H NMR (CDCl
3, 300 MHz) δ: 1.25 (t, 3 H, J = 7.1), 1.32 (t, 3 H, J = 7.1), 1.46 (s, 9 H), 3.47 (s (br), 1 H), 4.14–4.34 (m, 4H), 4.50 (s (br), 1 H), 4.83 (dd, 1 H, J = 7.9, 1.3), 5.51 (d, 1 H, J = 7.9);
13C NMR (CDCl
3, 50 MHz) δ:13.9, 14.0, 28.2, 56.9, 61.8, 62.1, 72.0, 80.3, 155.5, 168.5, 171.5; IR (film) cm
−1: 3450 (br), 2983, 1720, 1507, 1369, 1241, 1218, 1188, 1117, 1059, 1029.
33.
After isolation of the second crop, the recovered oily product mixture should be purified, if required, by column chromatography (100 g of silica gel/g oil) using
hexane–
ethyl acetate (8:1). The rate of crystallization for the third crop becomes much slower, requiring 2 to 3 days at −30°C; cooling at −78°C results in oil separation. Careful chromatography on silica gel of the oily residue remaining after concentration of the mother liquors of the third crystallization affords the pure 2R,3R diastereomer. Spectral data for
diethyl (2R,3R)-2-(N-tert-butoxycarbonyl)amino-3-hydroxysuccinate are as follows:
1H NMR (CDCl
3, 500 MHz) δ: 1.30 (t, 3 H, J = 6.9), 1.31 (t, 3 H, J = 7.0), 1.41 (s, 9 H), 3.45 (d, 1 H, J = 5.0), 4.22–4.30 (m, 4 H), 4.68 (d (br), 1 H, J = 3.3), 4.78 (d (br), 1 H, J = 8.3), 5.26 (d (br), 1H, J = 9.3);
13C NMR (CDCl
3, 50 MHz) δ: 13.9, 14.0, 28.0, 56.0, 61.9, 62.4, 71.1, 80.0, 155.2, 169.4, 171.9.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Optically active β-hydroxy-α-amino acids are important not only as constituents of biologically active peptides,
5 6 but also as precursors to β-lactam antibiotics.
7 8 9 10 Various synthetic approaches to compounds of this class have been reported involving the traditional resolution of racemic compounds,
11 asymmetric syntheses,
12 13 14 15 16 17 18 or the derivatization of appropriate chiral templates.
19 20 21 22 The procedure described here is a practical preparation of
diethyl erythro-3-hydroxy-N-(tert-butoxycarbonyl)-L-aspartate (
5) in high optical purity, amenable to large scale preparation. The synthetic route involves the transformation of
diethyl L-tartrate to diethyl
(2S,3S)-2-bromo-3-hydroxysuccinate (
2),
2 preparation of
diethyl (2R,3R)-2,3-epoxysuccinate (
3),
2 nucleophilic cleavage of the epoxide by azide to give
diethyl (2S,3R)-2-azido-3-hydroxysuccinate (
4),
9f and the single-pot transformation of the azido group into the N-(tert-butoxycarbonyl)amino group
23 to give
5 in high enantiomeric and diastereomeric purity.
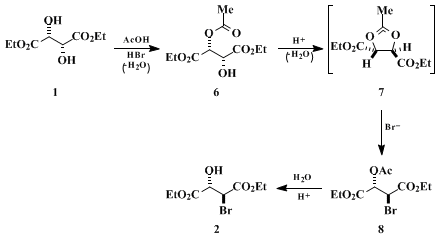
The first and second steps are examples of a standard method for the stereospecific synthesis of epoxides via vicinal acetoxy bromides from vicinal diols.
24 25 Based upon kinetic studies, the stereochemical outcome, and the observation of an intermediate carbocation by NMR spectroscopy,
24 the mechanism of the first step has been rationalized to involve monoacetylation of the diol, cyclization to a 1,3-dioxolan-2-ylium ion, and capture of this intermediate by bromide ion. If this mechanism is valid for the present case, the reaction can be illustrated as shown in Scheme 1. The experimental findings
(Note 3) correspond reasonably with this mechanistic scheme. Reaction of
1 with
30% HBr in acetic acid proceeded, as shown by both TLC and isolation experiments, with the initial formation of
8 (R
f = 0.53) followed by the gradual increase in the formation of
2 (R
f = 0.36) because of increase in the content of water liberated in the medium. At complete consumption of
1, an additional minor product
6 (R
f = 0.21) was detected by TLC. The presence of
6 may result from competition by the increasing concentration of water as the reaction progresses for
7 relative to bromide. Thus, although
1 had been completely consumed as observed by TLC, the subsequent transesterification process (ethanol/HCl) gives rise to a mixture of
2 and a small amount of
1 (derived from
6) which are easily separated by column chromatography on silica gel.
The optimal base for closure to epoxide 2 is sodium ethoxide in ethanol, as potassium hydroxide in ethanol, sodium or potassium carbonate in ethanol, or benzyltrimethylammonium hydroxide in ethanol resulted in the formation of a complex mixture of decomposition products arising mainly from deprotonation at carbon.
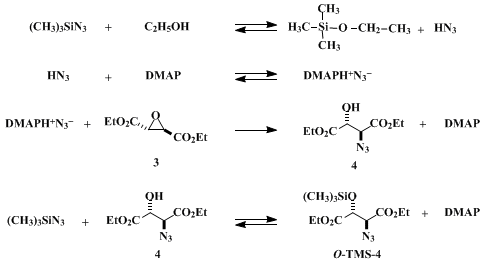
For the azide cleavage of chiral 2,3-epoxy diester
3, the submitter's earlier procedure
9f was modified to reduce the amount of azide reagent (
azidotrimethylsilane, TMSN
3) and lower the reaction temperature. This effort, coupled with the submitter's recent finding that such a reaction can be accelerated in the presence of amines,
18,
9i led to the use of the system described above. The reaction may proceed by the pathway shown in Scheme 2. The reactive azide species could be DMAPH + N
3− formed from DMAP and HN
3 which are, in turn, initially generated by the exothermic reaction of TMSN
3 and
ethanol, or later catalytically from reaction of TMSN
3 with the desired product
4. The reaction occurs with these reagents at a practical rate at 25°C, in sharp contrast to the previous more vigorous conditions (2 equiv or more of TMSN
3-CH
3OH (1:1) at 60°C in DMF).
9f In addition, this reaction does not require any special precautions other than use of a well-ventilated hood since the generation a high concentration of the hazardous, free HN
3 is avoided.
18,
9i The initial product is a mixture of
4 and its O-silylated derivative (O-TMS-
4). The latter is converted to
4 upon treatment of the mixture with
HCl in
ethanol affording
4 in over 85% yield.
The final one-pot, two-stage conversion of azide to an amino group and then to the related tert-butoxycarbonyl derivative was effected as before.
23 Catalytic hydrogenolysis of
4 over
10% Pd-carbon in ethyl acetate in the presence of
di-tert-butyl dicarbonate for 4 hr at room temperature provides a crude product consisting of
5 and its (2R,3R)-isomer (in the same ratio as that observed for
4 and its (2R,3R)-isomer) that can be separated by fractional crystallization from a
hexane–
ether mixed solvent at −30°C.
This procedure has been used by Ohfune
26 to convert N-(benzyloxycarbonyl)amino groups to N-(tert-butoxycarbonyl)amino groups in one pot, the Cbz to Boc switching protocol. This procedure is efficient because no tedious isolation of the intermediate amine is required. The following examples illustrate the advantages of this one-pot procedure. In addition, the amount of
palladium catalyst employed controls the chemoselectivity of the transformation. For instance, employing 1–2 wt% Pd/g of substrate, the 1,2-diazide
9 can be changed to the corresponding, protected 1,2-diamine derivative
10, while the O-benzyl group is kept unchanged.
9i On the other hand, employing 30 wt% Pd/g of substrate, both the azido and O-benzyl groups of
11 can be transformed to N-(tert-butoxycarbonyl)amino and hydroxyl groups, (
12) respectively (Scheme 3).
27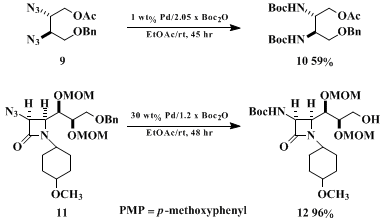
A recent report by Evans
28 of azide reduction by
SnCl2 in aqueous
dioxane and in situ amine protection by addition of Boc
2O has demonstrated that other methods of reduction of azides may be feasible if catalytic reduction of the azide is precluded because of the presence of protective groups sensitive to hydrogenolysis.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
brine
D-Aspartic acid, N-[(1,1-dimethylethoxy)carbonyl]-3-hydroxy-, diethyl ester, threo-
diethyl erythro-3-hydroxy-N-(tert-butoxycarbonyl)-L-aspartate
Pd-carbon
ethanol (64-17-5)
potassium carbonate (584-08-7)
hydrogen chloride,
HCl (7647-01-0)
acetic acid (64-19-7)
ethyl acetate,
EtOAc (141-78-6)
methanol (67-56-1)
ether (60-29-7)
hydrogen (1333-74-0)
acetyl chloride (75-36-5)
chloroform (67-66-3)
sodium bicarbonate (144-55-8)
sodium chloride (7647-14-5)
HYDROBROMIC ACID,
hydrogen bromide,
HBr (10035-10-6)
sodium sulfate (7757-82-6)
nitrogen (7727-37-9)
carbon (7782-42-5)
potassium hydroxide (1310-58-3)
sodium (13966-32-0)
sodium ethoxide (141-52-6)
palladium (7440-05-3)
L-tartaric acid (87-69-4)
triethyl orthoformate (122-51-0)
dichloromethane (75-09-2)
magnesium sulfate (7487-88-9)
dioxane (123-91-1)
N,N-dimethylformamide (68-12-2)
magnesium ethoxide (2414-98-4)
benzyltrimethylammonium hydroxide (100-85-6)
hexane (110-54-3)
calcium hydride (7789-78-8)
hydrogen azide
azidotrimethylsilane (4648-54-8)
diethyl L-tartrate
4-(dimethylamino)pyridine (1122-58-3)
Diethyl (2S,3S)-2-bromo-3-hydroxysuccinate (80640-14-8)
Diethyl (2R,3R)-2,3-epoxysuccinate (74243-85-9)
diethyl (2S,3R)-2-bromo-3-hydroxy succinate
Diethyl (2S,3R)-2-azido-3-hydroxysuccinate (101924-62-3)
Diethyl (2S,3R)-2-(N-tert-butoxycarbonyl)amino-3-hydroxysuccinate,
DIETHYL (2S,3R)-2-(N-tert-BUTOXYCARBONYL)AMINO- 3-HYDROXYSUCCINATE (174682-54-3)
ethoxytrimethylsilane (1825-62-3)
diethyl (2S,3R)-2-azido-3-(trimethylsiloxy)succinate
diethyl (2R,3R)-2-(N-tert-butoxycarbonyl)amino-3-hydroxysuccinate
Di-tert-butyl dicarbonate (24424-99-5)
(2S,3S)-2-bromo-3-hydroxysuccinate
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved