Org. Synth. 1993, 71, 39
DOI: 10.15227/orgsyn.071.0039
DIRECT DEGRADATION OF THE BIOPOLYMER POLY[(R)-3-HYDROXYBUTYRIC ACID] TO (R)-3-HYDROXYBUTANOIC ACID AND ITS METHYL ESTER
[Butanoic acid, 3-hydroxy-, (R)-, homopolymer to Butanoic acid, 3-hydroxy-, (R)- and Butanoic acid, 3-hydroxy-, methyl ester, (R)-]
Submitted by Dieter Seebach, Albert K. Beck, Richard Breitschuh, and Kurt Job
1.
Checked by Eugene R. Hickey and Leo A. Paquette.
1. Procedure
A. (R)-(−)-Methyl 3-hydroxybutanoate. A 2-L, round-bottomed flask is charged with 50 g (0.58 mol) of poly-[(R)-3-hydroxybutyric acid] (PHB) (Note 1) and 500 mL of absolute 1,2-dichloroethane. The flask is equipped with a reflux condenser, and the mixture is heated at reflux for 1 hr. A solution of 10 mL of concd sulfuric acid in 200 mL of absolute methanol is added and the reaction mixture is heated at reflux for 3 days. During this time the mixture becomes homogeneous.
After the reaction mixture is cooled to room temperature, 100 mL of half-saturated brine is added to the reaction mixture, which is stirred for 10 min before the layers are separated. The aqueous layer is extracted three times with 200 mL each of chloroform. The combined organic layers are washed with 100 mL of brine, 100 mL of saturated sodium bicarbonate solution, and 100 mL of brine. After the solution is dried over magnesium sulfate and removed in a rotary evaporator, a residue of 59 g is isolated.
This crude product is distilled under reduced pressure to give 48 g (70%) of pure (R)-(−)-methyl 3-hydroxybutanoate (bp 61–62°/18 mm) with a specific rotation of [α]DRT −47.6° (CHCl3, c 1.0) (Note 2).
B. (R)-(−)-3-Hydroxybutanoic acid. In a 1-L, two-necked, round-bottomed flask, fitted with a precision-ground stirrer and a coil condenser bearing a distillation bridge, are placed 200 mL of 1,2-dichloroethane and 50 g (0.58 mol) of PHB (Note 1). The mixture is stirred and heated at 150°C until a milky solution is formed (Note 3). p-Toluenesulfonic acid monohydrate (2.5 g, 0.013 mol) is added and the solution heated an additional 30 min at 150°C. A total amount of 9.3 g (11.8 mL, 0.29 mol) of methanol is then added carefully through the condenser to the boiling solution in approx 1-mL portions (Note 4). The light brown, cloudy solution is heated at reflux (oil bath, 140°C) for another 48 hr (Note 5). The oil bath temperature is reduced to 130°C and 60 mL of water is carefully added. The resulting two-phase mixture is heated for another 4 hr (Note 6). Methanol, 1,2-dichloroethane, and approx 150 mL of water are removed by distillation through the condenser within 1–2 hr (Note 7). During the distillation nine 30-mL portions of water are added; the resulting homogeneous aqueous solution is heated at reflux for another 4 hr. During this period the temperature of the oil bath is increased to 180°C. Methanol/water (50 mL) is removed by distillation and, after the addition of 50 mL of water (Note 8), the solution is again heated at reflux for 15 hr. The reaction mixture is cooled to about 30°C, followed by the addition of 0.6 g (0.015 mol) of sodium hydroxide in 10 mL of water. Water is first removed in a rotary evaporator at a maximum temperature of 30°C, 15 mm, followed by drying at 0.1 mm (Note 9). The residue is dissolved in 250 mL of ether, the solution is dried over magnesium sulfate, filtered, concentrated in a rotary evaporator and dried at 0.1 mm. The resulting light-brown clear oil (Note 10) is distilled at 80–100°C (air bath)/10−3mm by bulb to bulb distillation (Note 11). The crude product is either crystalline or a colorless oil that crystallizes upon standing at room temperature. After the crystals are crushed in a mortar, they are dried for 2 days in a desiccator over phosphorus pentoxide (P2O5) (Note 12), to yield 32 g (53%) of (R)-3-hydroxybutanoic acid, mp 44–46°C, [α]DRT −24.7° (H2O, c 5.0 (Note 13), (Note 14), (Note 15).
2. Notes
1.
The submitters used
PHB=Poly-(R)-3-hydroxybutyric acid obtained from Marlborough Biopolymers Ltd., Rudby Hall, Hutton Rudby, Yarm, Cleveland, England, PHB homopolymer, BX GO4, technical grade powder, MBL 100/703. The checkers used
PHB from Fluka Chemie AG, CH-8470 Buchs, Switzerland. The submitters report that the procedure works well on batch sizes of 200–250 g.
2.
The spectroscopic properties are as follows: IR (neat) cm
−1: 1735 (C=O);
1H NMR (90 MHz, CDCl
3) δ: 1.25 (d, 3 H, CH
3), 2.45 (d, 2 H, CH
2), 3.00 (br s,1 H, OH), 3.75 (s, 3 H, OCH
3), 4.20 (m, 1 H, CH). For further data see ref.
2.
3.
The submitters used an oil bath to insure vigorous reflux. The time required to obtain the desired solution consistency depends on the PHB used (30 min to over 2 hr).
4.
The reaction solution should be maintained at a vigorous reflux. Should the solution cool during the addition a white flaky precipitate may form that redissolves upon extended heating (up to 4 hr at 140°C).
5.
This results in a mixture of oligomeric methyl esters with an average degree of polymerization of 2.1–2.3, according to
1H NMR analysis (ratio of the CH
3C-signals vs. CH
3O-signals of the esters).
6.
Most of the methyl ester groups are saponified with release of
methanol.
7.
It is crucial that all the
methanol and
1,2-dichloroethane be removed from the reaction mixture to insure that all the oligomers are in the aqueous phase.
8.
Newly formed
methanol is removed at this stage. Only a small amount of methyl esters remains in the mixture at this point.
9.
Low temperature during evaporation avoids condensation back to oligomers.
10.
According to
1H NMR analysis the crude product consists of more than
93% monomeric
(R)-3-hydroxybutanoic acid, contaminated by
1–2% of
methyl (R)-3-hydroxybutanoate and less than
0.2% of crotonic acid.
11.
The crude product is placed in a 1-L, round-bottomed flask and distilled in an Aldrich Kugelrohr apparatus, heated with an
air bath, the receiver bulb being cooled with ice water. Partial condensation of
(R)-3-hydroxybutanoic acid to oligomers occurs if the temperature is too high during distillation
(Note 12).
12.
Drying in an
evacuated desiccator over
P2O5 removes water as well as
methyl (R)-3-hydroxybutanoate and
crotonic acid.
13.
The spectra are as follows: IR (neat) cm
−1: 1710 (C=O);
1H NMR (90 MHz, CDCl
3) δ: 1.25 (d, 3 H, CH
3), 2.50 (d, 2 H, CH
2), 4.20 (m, 1 H, CH), 7.70 (br s, 2 H, OH). For further data see ref.
2.
14.
The distillation residue (8–9 g) consists of water-soluble oligomers that can be saponified without loss of optical purity by heating at reflux with aqueous acid for 15 hr.
15.
The submitters report a yield of
45–48 g (
74.5–79%) using
PHB obtained from Marlborough Biopolymers. The checkers verified this result using
PHB obtained from the submitters. The yield reported here represents the checker's results with
PHB obtained from Fluka Chemie.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Optically active
3-hydroxybutanoic acid and its methyl ester were first prepared by McKenzie, Magnus-Levy, and Emil Fischer.
3 The biopolymer
PHB and mixed polymers containing
(R)-3-hydroxybutanoate and
(R)-3-hydroxypentanoate were also discovered long ago,
4,5 and are now produced on an industrial scale.
6,7 8 As described here, depolymerization by transesterification [H
+ or
Ti(OR)4 catalysis], or by hydrolysis, produces
9,10 11 12 the corresponding monomeric (R)-esters and (R)-acids
1. The
3-hydroxybutanoic acid can also be prepared by hydrolysis of the ester.
2,13The enantiomeric compounds, ent-
1, are available from
1 by inversion through sulfonates
14 or through β-lactones.
15 16 Ethyl (S)-3-hydroxybutanoate of 86–95 % ee has also been made by yeast reduction of acetoacetate.
17 Either the ester
1 or ent-
1 can be prepared by enantioselective hydrogenation of the
methyl acetoacetate with a
Ru-BINAP catalyst.
18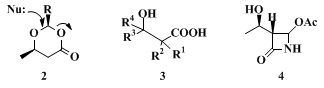
The hydroxy acid derivatives
1 are most versatile auxiliaries and building blocks for the synthesis of enantiomerically pure products.
7,11,19 Thus, in an improvement of Johnson´s method,
20 the dioxanones
2 can be used for overall enantioselective nucleophilic additions to aldehydes.
21 Through open-chain
22 or cyclic
23 lithium enolates derived from
1 and, through dioxinones,
24 new substituents can be introduced at carbons (2), (3) and (4) of hydroxybutanoic or
hydroxypentanoic acid to give enantiomerically pure products
25,26 of type
3. Another application is the incorporation of
(R)-3-hydroxybutanoic acid into the antibiotic thienamycin
27 through the β-lactam
4.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
H2O
P2O5
brine
(R)-(−)-Methyl 3-hydroxybutanoate
(R)-(−)-3-Hydroxybutanoic acid
PHB
POLY[(R)-3-HYDROXYBUTYRIC ACID]
homopolymer to Butanoic acid, 3-hydroxy-, (R)-
poly-[(R)-3-hydroxybutyric acid]
sulfuric acid (7664-93-9)
methanol (67-56-1)
ether (60-29-7)
sodium hydroxide (1310-73-2)
chloroform (67-66-3)
sodium bicarbonate (144-55-8)
1,2-dichloroethane (107-06-2)
magnesium sulfate (7487-88-9)
crotonic acid (3724-65-0)
Methyl acetoacetate (105-45-3)
3-Hydroxybutanoic acid (300-85-6)
phosphorus pentoxide (1314-56-3)
p-toluenesulfonic acid monohydrate (6192-52-5)
(R)-3-Hydroxybutanoic acid,
Butanoic acid, 3-hydroxy-, (R)- (625-72-9)
methyl (R)-3-hydroxybutanoate,
Butanoic acid, 3-hydroxy-, methyl ester, (R)-,
(R)-3-hydroxybutanoic acid, methyl ester (3976-69-0)
(R)-3-hydroxybutanoate
(R)-3-hydroxypentanoate
Ethyl (S)-3-hydroxybutanoate (56816-01-4)
hydroxypentanoic acid (13392-69-3)
(S)-3-hydroxybutanoic acid (6168-83-8)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved