Org. Synth. 1996, 73, 94
DOI: 10.15227/orgsyn.073.0094
REGIO- AND STEREOSELECTIVE INTRAMOLECULAR HYDROSILYLATION OF α-HYDROXY ENOL ETHERS: 2,3-syn-2-METHOXYMETHOXY-1,3-NONANEDIOL
[1,3-Nonanediol, 2-(methoxymethoxy)-, (R,R)-(±)-]
Submitted by Kohei Tamao
1, Yoshiki Nakagawa
2, and Yoshihiko Ito
2.
Checked by Michael R. Reeder, Lisa M. Reeder, and Robert K. Boeckman, Jr..
1. Procedure
A. 2-Bromoethyl methoxymethyl ether. A 1-L, single-necked, round-bottomed flask is equipped with a magnetic stirring bar and a gas inlet valve (Note 1). The flask is purged with argon, and charged with 125.0 g of 2-bromoethanol (1.00 mol), and 500 mL of dimethoxymethane (Note 2) and (Note 3). The resulting mixture is cooled to 0°C in an ice-water bath. As a single portion, 71.0 g (0.5 mol) of solid phosphorus pentoxide (P2O5) is added with stirring (Note 4). The resulting mixture, which becomes quite viscous because of the insoluble P2O5, is stirred at 0°C for 10 min, then warmed to room temperature and stirred for an additional 7 hr during which time the viscosity of the mixture decreases. The mixture is transferred to a 2-L separatory funnel, 200 mL of water is added, and the resulting mixture is extracted three times with 50 mL of ether. The combined organic extracts are washed successively with 100 mL of water and 100 mL of saturated aqueous sodium carbonate solution, dried over magnesium sulfate, filtered, and carefully concentrated with a rotary evaporator under reduced pressure (20–30 mm) at room temperature. The residue is distilled under reduced pressure to afford 131.3 g (78%) of 2-bromoethyl methoxymethyl ether as a colorless liquid, bp 74–78°C (40 mm) (Note 5).
B. Methoxymethyl vinyl ether. A 200-mL, two-necked, round-bottomed flask is equipped with a stopper, magnetic stirring bar (Note 6), and an efficient reflux condenser that is attached through a thick rubber tube to a clean trap, cooled in a dry ice-acetone bath, whose outlet is equipped with a calcium chloride drying tube. The flask is charged with 74.2 g (440 mmol) of 2-bromoethyl methoxymethyl ether, 58 g (880 mmol) of 85% potassium hydroxide pellets (Note 7), and 7.09 g (2.2 mmol) of tris[2-(2-methoxyethoxy)ethyl]amine (TDA-1) (Note 8). The mixture is heated to 140°C with stirring. The clear liquid phase gradually becomes cloudy and begins to reflux. After heating for 27 hr, consumption of the starting material is confirmed by GLC (Note 9). The calcium chloride tube is replaced with an additional cold trap that is attached to a water aspirator. The reaction mixture is cooled to 110°C and a vacuum is applied using the water aspirator for 2 hr, during which time volatile material is collected in the first cold trap. After the flask is cooled to room temperature, the first trap is disconnected and allowed to warm to room temperature. The liquid is transferred to a 50-mL, round-bottomed flask. A small amount of water (about 1 mL) is usually present and is removed with a syringe. The organic layer is distilled at atmospheric pressure into an ice-cooled receiver to afford 26.6–28.2 g (69–72%) of methoxymethyl vinyl ether as a colorless liquid, bp 64–70°C (Note 10). The product is dried and kept over 4 Å molecular sieves.
C. 2-Methoxymethoxy-1-nonen-3-ol. A 500-mL, two-necked, round-bottomed flask is equipped with a rubber septum, a magnetic stirring bar, and a pressure-equalizing dropping funnel bearing a three-way stopcock connected to a mineral oil bubbler. The flask is evacuated, purged with nitrogen, and charged with 200 mL of dry tetrahydrofuran (THF) (Note 11) and 8.83 g of methoxymethyl vinyl ether (100 mmol). The solution is cooled to −78°C with a dry ice-acetone bath, and then 88.7 mL (100 mmol) of sec-butyllithium (1.13 M in cyclohexane) is added dropwise over 15 min with stirring via syringe. After the addition is complete, the yellow mixture is allowed to warm to −30°C over 30 min and stir for 1 hr. The mixture is recooled to −78°C and 11.44 g (100 mmol) of heptanal is added dropwise over 5 min through the dropping funnel. The reaction mixture is allowed to warm to room temperature over 30 min and to stir for 3 hr. A 100-mL portion of saturated aqueous sodium chloride solution is added, the mixture is transferred to a 1-L separatory funnel, mixed thoroughly, and the organic layer separated. The aqueous layer is extracted three times with 50 mL of diethyl ether. The combined organic phases are dried over sodium sulfate (Na2SO4), filtered, and concentrated under reduced pressure with a rotary evaporator. The residue is distilled under reduced pressure to afford 12.6–13.1 g (62–65%) of 2-methoxymethoxy-1-nonen-3-ol as a colorless liquid, bp 82–84°C (0.4 mm) (Note 12).
D. 2-Methoxymethoxy-1,3-nonanediol. A 500-mL, single-necked, round-bottomed flask, equipped with a three-way stopcock connected to a bubbler as above and a magnetic stirring bar, is charged with 14.79 g (73.1 mmol) of 2-methoxymethoxy-1-nonen-3-ol (Note 6). The flask is evacuated, then purged with nitrogen, and charged with 350 mL of dry diethyl ether and 8.88 g (87.7 mmol) of triethylamine (Note 13). After the mixture is cooled to 0°C in an ice-water bath, 8.30 g (87.7 mmol) of chlorodimethylsilane is added slowly with stirring. A large quantity of salt appears immediately. The white suspension is stirred at room temperature for 1 hr. The resulting mixture is then filtered through a sintered-glass Büchner funnel and the filter cake is washed thoroughly with dry hexane (Note 14). The combined filtrate and washings are concentrated under reduced pressure with a rotary evaporator. Some salts usually remain in the residue and are removed by dilution of the residue with an additional 50 mL of dry hexane followed by filtration, washing the filter cake with hexane, and concentration of the combined filtrate and washings as above. This procedure for removal of salts is repeated two to three times until the colorless residual liquid remains clear after concentration (Note 15). The residual liquid is finally subjected to high vacuum (0.4 mm) for 2 hr to complete removal of volatile material, and then transferred to a 300-mL, round-bottomed flask, equipped with a three-way stopcock and a magnetic stirring bar. The flask is evacuated, purged with nitrogen, and charged with 73 mL of dry hexane. To this mixture is added 1.21 mL (0.35 mmol) of the previously prepared platinum catalyst solution (Note 16) at room temperature with stirring. An exothermic reaction ensues almost immediately and the temperature of the mixture rises to about 30°C in 15 min, during which time the color of the clear mixture changes to yellow. The exothermic reaction ceases in about 1 hr. After the reaction mixture is stirred for an additional 1.5 hr at room temperature, completion of the reaction is confirmed by analysis by 1H NMR (Note 17). A 7.3-g portion of activated carbon powder is then added to the lightly colored mixture to effect removal of organoplatinum species (Note 18). The black suspension is stirred for about 12 hr, and suction filtered through Celite into a 500-mL, two-necked, round-bottomed flask. The filter cake is washed with dry hexane, and the combined filtrate and washings are concentrated by rotary evaporation (Note 19). The flask containing the residue is equipped with a magnetic stirring bar and a thermometer, and is kept open to the air throughout subsequent manipulations. The flask is charged with 73 mL of THF, 73 mL of methanol (Note 20), 7.32 g (73.1 mmol) of solid potassium hydrogen carbonate, and 8.5 g (146 mmol) of solid potassium fluoride (Note 21). To the stirred mixture is added 29.7 mL (263 mmol) of 30% hydrogen peroxide slowly at room temperature (Note 22). A somewhat cloudy upper organic layer and a milky white lower aqueous layer result. After several minutes, an exothermic reaction begins which is controlled by intermittent, brief cooling with a water bath to maintain the temperature at about 40°C. The exothermic reaction ceases in about 2 hr. The mixture is stirred at room temperature for an additional 3 hr (Note 23). The excess hydrogen peroxide is decomposed by careful dropwise addition of a 50% aqueous sodium thiosulfate pentahydrate solution (~20 mL) with stirring over 20 min during which time the temperature is maintained near 30°C by intermittent cooling with an ice bath (Note 24). A negative starch-iodide test is observed after addition is complete (Note 25). The mixture is concentrated by rotary evaporator at 40–50°C under reduced pressure (water aspirator). To the residual aqueous suspension is added 150 mL of anhydrous ethanol and the mixture is stirred vigorously until the sticky, pale yellow precipitate becomes granular. The mixture is concentrated as above to give a pale yellow solid. The ethanol addition and evaporation procedure is repeated once more to insure thorough removal of water. The residue is then diluted with 150 mL of diethyl ether, the mixture is filtered with suction, and the filter cake is washed well with two 100-mL portions of diethyl ether. The combined filtrate and washings are concentrated by rotary evaporation. The residual, crude product is purified by chromatography on 250 g of silica gel with hexane/ethyl acetate (1:1 v/v) as eluent to give 9.34–10.62 g (58–66%) of 2-methoxymethoxy-1,3-nonanediol (Rf = 0.2) as a colorless liquid (Note 26). The syn and anti isomers cannot be separated by column chromatography. However, separation can be effected by GLC which showed the isomer ratio to be 98:2 syn/anti (Note 27).
2. Notes
1.
A powerful magnetic stirrer and a large rugby ball-shaped stirring bar can be used. The checkers suggest use of an overhead mechanical stirrer because of the viscosity of the mixture in the initial stages of the reaction, which makes efficient magnetic stirring difficult.
2.
2-Bromoethanol was obtained from Aldrich Chemical Company, Inc., and was distilled prior to use.
3.
Dimethoxymethane was purchased from the Aldrich Chemical Company, Inc., and used as received.
4.
Reagent grade
phosphorus pentoxide was obtained from the J. T. Baker Chemical Company and used as received.
5.
2-Bromoethyl methoxymethyl ether exhibits the following spectral properties:
1H NMR (200 MHz, CDCl
3) δ: 3.38 (s, 3 H), 3.49 (t, 2 H, J = 6.2), 3.85 (t, 2 H, J = 6.2), 4.66 (s, 2H);
13C NMR (50 MHz, CDCl
3) δ: 30.74, 55.45, 67.79, 96.54; IR (neat) cm
−1: 2952, 2900, 1148, 1118, 1072, 1036. Anal. Calcd for C
4H
9BrO
2: C, 28.43; H, 5.37. Found: C, 28.39; H, 5.36.
6.
A
powerful magnetic stirrer and a large rugby ball-shaped stirring bar can be used. The checkers suggest use of an overhead mechanical stirrer because of the large quantity of salts produced in the reaction, which makes efficient magnetic stirring difficult.
7.
Potassium hydroxide pellets are better than finely ground powder, because the powder readily solidifies into a mass upon heating causing the reaction to slow down. Sonication does not facilitate the reaction.
8.
TDA-1 is not essential, but accelerates the reaction considerably.
9.
Isothermal GLC analysis was performed on a 30-m × 0.32-mm ID HP-5 capillary column containing 5% phenylmethylsilicone as a stationary phase at 50°C (retention time of the
bromo ether is 3.8 min).
10.
Methoxymethyl vinyl ether exhibits the following spectral properties:
1H NMR (200 MHz, C
6D
6) δ: 3.11 (s, 3 H), 4.09 (dd, 1 H, J = 6.6, 1.4), 4.54 (s, 2 H), 4.61 (dd, 1 H, J = 14.2, 1.4), 6.32 (dd, 1 H, J = 14.2, 6.6);
13C NMR (50 MHz, C
6D
6) δ: 55.37, 90.71, 95.28, 149.94.
11.
Tetrahydrofuran was distilled from
sodium benzophenone ketyl.
12.
Since
2-methoxymethoxy-1-nonen-3-ol tends to decompose on attempted GLC analysis, the purity is best checked by
1H NMR.
2-Methoxymethoxy-1-nonen-3-ol exhibits the following spectral properties:
1H NMR (200 MHz, C
6D
6) δ: 0.91 (t (br), 3 H, J = 7), 1.20–1.95 (m, 11 H), 3.17 (s, 3 H), 4.02 (q (br), 1 H, J = 5), 4.35 (s, 2 H), 4.75 (d, 1H, J = 6), 4.78 (d, 1 H, J = 6);
13C NMR (50 MHz, C
6D
6) δ: 14.27, 22.99, 25.88, 29.64, 32.20, 35.73, 55.71, 72.95, 84.21, 93.87, 162.63; IR (neat) cm
−1: 3448, 2940, 2864, 1644, 1156, 1096, 1020. Anal. Calcd for C
11H
22O
3: C, 65.31; H, 10.96. Found: C, 65.48; H, 11.17.
13.
Triethylamine was distilled from
calcium hydride. Freshly distilled
triethylamine should be used, because any impurities in aged
triethylamine may poison the catalyst in the next step.
14.
Hexane was distilled from
sodium.
15.
Since the O-silylated product is somewhat sensitive to moisture, the filtration process should be performed quickly.
16.
Platinum catalyst preparation: a mixture of
1 g of chloroplatinic acid hexahydrate,
12.4 mL (22 equiv) of 1,3-divinyltetramethyldisiloxane (an excess), and 1.2 mL of water is heated to 65–70°C for 3.5 hr with stirring.
3 4 The initial mixture becomes homogeneous after ~30 min. The solution is then cooled to room temperature, and
5 mL of aqueous saturated sodium bicarbonate (NaHCO3) solution is added. The upper, yellow-orange, organic layer was separated, dried over Na
2SO
4, and filtered affording a solution (~0.29 M) of the
platinum catalyst in disiloxane that was stored in the refrigerator until use.
17.
An aliquot is taken into an NMR tube, solvent is removed under reduced pressure, C
6D
6 is added and the sample is analyzed by
1H-NMR. The following spectral and analytical data for the cyclic siloxanes were obtained. The syn/anti ratio (95/5) was determined by capillary GLC
(Note 9) except that a temperature program was employed [100°C for 2 min, then increased 15°C/min to 250°C (retention times 6.46 min (anti), 6.66 min (syn)]. Each isomer was isolated by preparative GLC: Syn:
1H NMR (200 MHz, C
6D
6) δ: 0.17 (s, 3 H), 0.29 (s, 3 H), 0.78 (dd, 1 H, J = 15, 5.5), 0.92 (t (br), 3 H, J = 7), 1.03 (dd, 1 H, J = 15, 5), 1.20–1.65 (m, 7 H), 1.65–2.00 (m, 3 H), 3.23 (s, 3 H), 3.89 (dt (br), 1 H, J = 9, 4), 4.12 (q (br), 1 H, J = 5), 4.48 (d, 1 H, J = 7), 4.65 (d, 1 H, J = 7);
13C NMR (50 MHz, C
6D
6) δ: 0.28, 1.14, 14.30, 18.39, 23.04, 26.72, 29.96, 32.18, 32.33, 55.28, 77.91, 80.91, 95.16; IR (neat) cm
−1: 2963, 2864, 1470, 1254, 1150, 1102, 1046, 872. Anti:
1H NMR (200 MHz, C
6D
6) δ: 0.15 (s, 3 H), 0.23 (s, 3 H), 0.83 (dd, 1 H, J = 14.2, 7.8), 0.91 (t (br), 3 H, J = 6 ), 1.18 (dd, 1 H, J = 14, 6), 1.25–1.85 (m, 10 H), 3.26 (s, 3 H), 3.90–4.14 (m, 2 H), 4.57 (d, 1 H, J = 7), 4.73 (d, 1 H, J = 7). Anal. Calcd for C
13H
28O
3Si (isomeric mixture): C, 59.95, H, 10.84. Found: C, 59.67; H, 11.02.
18.
It is essential to remove all organoplatinum species since these species rapidly decompose
hydrogen peroxide in the next oxidation step.
19.
The filtrate is ordinarily colorless but may possibly be brown, owing to the presence of trace amounts of organoplatinum species.
20.
Commercial
reagent grade THF and methanol are used without further purification.
21.
Reagent grade anhydrous
potassium fluoride was purchased from J. T. Baker Chemical Company. This material must be weighed quickly because it is highly hygroscopic.
22.
Slow, not vigorous, evolution of small bubbles of
oxygen due to a trace amount of residual organoplatinum species may be observed.
23.
Since the hydrosilylation products decompose on
silica gel, consumption of the starting material cannot be monitored by TLC. Only the formation of the product diol can be detected (R
f = 0.2,
hexane/
ethyl acetate 1:1).
24.
CAUTION: The thiosulfate solution MUST NOT be added in one portion; since a sudden, violent, uncontrollable reaction could occur.25.
If the starch-iodide test is still positive for the presence of
peroxide, additional
thiosulfate solution should be added until a negative starch-iodide test is obtained.
26.
The major syn isomer of
2-methoxymethoxy-1,3-nonanediol exhibits the following spectral properties:
1H NMR (200 MHz, CDCl
3) δ: 0.86 (t (br), 3 H, J = 7), 1.18–1.50 (m, 10 H), 2.52 (d, 1 H, J = 5), 2.95 (dd, 1 H, J = 8, 4), 3.35–3.50 (m, 4 H, including a sharp singlet at 3.44), 3.55–3.82 (m, 3 H), 4.71 (d, 1 H, J = 7), 4.80 (d, 1 H, J = 7);
13C NMR (50 MHz, CDCl
3) δ: 14.02, 22.54, 25.43, 29.23, 31.72, 33.31, 55.89, 63.22, 71.80, 84.12, 97.43; IR (neat) cm
−1: 3428, 2936, 1470, 1154, 1108, 1032. Anal. Calcd for C
11H
24O
4 (isomer mixture): C, 59.97; H, 10.98. Found: C, 59.78; H, 11.04.
27.
The syn/anti ratio (98:2) was determined by capillary GLC on a
30-m × 0.32-mm ID HP-5 capillary column containing 5% phenylmethylsilicone as the stationary phase using the following temperature program: 100°C for 2 min, then increase 15°C/min to 250°C (retention times 6.96 min (anti), 7.55 min (syn)). The submitters report that the syn/anti ratio can be determined after conversion to the acetonide by treatment with excess
dimethoxypropane and catalytic acid. By this procedure, the isomer ratio was found to be 94.4:5.6 (syn/anti). The submitters also report that the isomers can be separated easily by chromatography on silica gel: R
f = 0.31 for the syn isomer and R
f = 0.45 for the anti isomer (
hexane/
ethyl acetate 1:1). The acetonides exhibit the following spectral properties: syn:
1H NMR (200 MHz, CDCl
3) δ: 0.85 (t (br), 3 H, J = 7), 1.15–1.75 (m, 16 H, including two sharp singlets at 1.41 and 1.42), 3.33 (q, 1 H, J = 2.0), 3.39 (s, 3 H), 3.80–4.04 (m, 3 H), 4.64 (d, 1 H, J = 7.1), 4.78 (d, 1 H, J = 7.1);
13C NMR (50 MHz, CDCl
3) δ: 14.01, 18.89, 22.53, 24.92, 29.17, 31.05, 31.72, 55.71, 62.82, 70.00, 71.30, 95.29, 98.52 (Only five signals are observed for the hexyl group). Anal. Calcd for C
14H
28O
4: C, 64.58; H, 10.84. Found: C, 64.42; H, 11.11. Anti:
1H NMR (200 MHz, CDCl
3) δ: 0.81 (t (br), 3 H, J = 6.5), 1.10–1.80 (m, 16 H, including two sharp singlets at 1.30 and 1.38), 3.23–3.38 (m, 4 H, including a sharp singlet at 3.29), 3.50–3.68 (m, 2 H), 3.90 (dd, 1 H, J = 11.4, 5.2), 4.54 (d, 1 H, J = 6.8), 4.59 (d, 1 H, J = 6.8);
13C NMR (50 MHz, CDCl
3) δ: 14.08, 19.76, 22.62, 25.06, 28.23, 29.24, 31.83, 32.34, 55.62, 63.39, 72.11, 74.48, 96.49, 98.60. The checkers found the determination of the syn/anti ratio by this procedure to be unreliable because of the presence of interfering impurities formed during conversion to the acetonide.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
This procedure for the preparation of
2-methoxymethoxy-1,3-nonanediol is a modification of that reported by the submitters.
5 Among a wide variety of methodologies for stereoselective construction of polyoxygenated skeletons, the most straightforward is the stereoselective introduction of two
oxygen functionalities onto carbon-carbon double bonds, as represented by the Sharpless epoxidation
6 or osmium tetroxide oxidation of allyl alcohols.
7 An alternative route may be envisioned by anti-Markovnikov hydration of enol ether derivatives, as in the present method, but such an approach has so far been rarely studied. The only other reported method is hydroboration which affords syn/anti ratios in the range of 83/17 to 3/97 in essentially the same systems as those examined in this procedure.
8 The method described here is a highly stereoselective route to 2,3-syn isomers of 1,2,3-triol skeletons from α-hydroxy enol ethers.
This procedure consists of the synthesis of a precursor,
methoxymethyl vinyl ether, an α-hydroxy enol ether, and the intramolecular hydrosilylation of the latter followed by oxidative cleavage of the silicon-carbon bonds. The first step, methoxymethylation of
2-bromoethanol, is based on Fujita's method.
9 The second and third steps are modifications of results reported by McDougal and his co-workers.
10 Dehydrobromination of
2-bromoethyl methoxymethyl ether to
methoxymethyl vinyl ether10 was achieved most efficiently with
potassium hydroxide pellets
10,11 rather than with
potassium tert-butoxide as originally reported for dehydrobromination of the tetrahydropyranyl analog.
12 Potassium tert-butoxide was effective for the dehydrobromination, but formed an adduct of tert-butyl alcohol with the vinyl ether as a by-product in substantial amounts.
Methoxymethyl vinyl ether is lithiated efficiently with
sec-butyllithium in THF
10 and, somewhat less efficiently, with
butyllithium in
tetrahydrofuran. Since lithiation of simple vinyl ethers such as
ethyl vinyl ether requires
tert-butyllithium,
13 metalation may be assisted by the methoxymethoxy group in the present case.
In the final step for this large scale preparation, the hydroxyl group is silylated with
chlorodimethylsilane and
triethylamine to effect introduction of the dimethylsilyl group. Alternatively,
(diethylamino)dimethylsilane (HMe2SiNEt2) or
1,1,3,3-tetramethyldisilazane (HMe2Si)2NH in the presence of a catalytic amount of
ammonium chloride as a promoter can also be used. In these cases, the filtration step can be omitted since removal of the generated amine and the excess silylamine affords the crude oily product that can be directly employed in the hydrosilylation step.
5,14 Intramolecular hydrosilylation of α-hydroxy enol ethers proceeds readily with a neutral
platinum catalyst such as the
vinylsiloxane/
platinum(0) complex
3,4 since the most commonly used catalyst,
chloroplatinic acid hydrate (H2PtCl6·6H2O), is not suitable for use with acid-sensitive enol ethers. Similar regio- and stereoselective intramolecular hydrosilylations can also be achieved with rhodium catalysts.
15 For removal of organoplatinum species prior to the oxidation, activated
carbon seems to be more efficient than the
disodium salt of ethylenediaminetetraacetic acid (EDTA·2Na) originally used.
5 Oxidative cleavage of the silicon-carbon bond is performed under standard conditions.
16 17 18Finally, the regio- and stereoselectivities of the intramolecular hydrosilylation of allyl alcohols, homoallyl alcohols, and allylamines are summarized in Scheme 1. While these results have been obtained with the dimethylsilyl (HMe2Si) group in the presence of a platinum catalyst, stereoselectivity, but not regioselectivity, can be controlled by other silyl groups such as the diphenyl (HPh2Si) or di(isopropyl)silyl [H(i-Pr)2Si] groups.
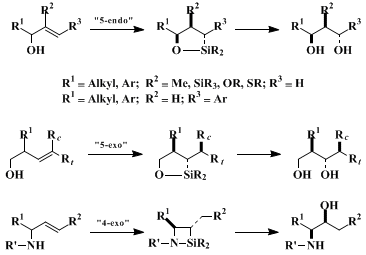
In the allylic alcohol series having a terminal olefin, the reaction proceeds in the 5-endo fashion to give five-membered ring compounds selectively with the syn stereoisomer being predominant, regardless of the nature of the catalyst,
platinum or rhodium.
19 20,21 Stereoselectivity increases with an increase in bulk of the allylic substituent. In addition, much higher selectivities are attained with enol ethers (R
2 = OMOM, OTHP, SR)
5 in comparison to other systems [(R
2 = Me, SiMe
2(O-i-Pr)
22]. Stereoselectivities in cyclic enol ethers appear to be sensitive to the reaction conditions.
23 It has also been shown that the 5-endo cyclization proceeds smoothly with allyl alcohols with a terminal aryl group (R
3 = Ar), but not with an alkyl group.
24 In
homoallyl alcohols, the 5-exo type of intramolecular hydrosilylation proceeds to form five-membered cyclic compounds and eventually 1,3-diols by oxidative cleavage. Two types of 1,2-stereoselection are possible, one being anti-controlled by the allylic substituent and, the other depending upon the olefin geometries, anti to the cis substituent R
c and syn to the trans R
t.
19,20 Catalytic, asymmetric, intramolecular hydrosilylation is also possible with rhodium catalysts in the presence of optically active phosphine ligands.
15,24 In contrast, allylamines form four-membered cyclic products via 4-exo ring closure in the platinum-catalyzed hydrosilylation from which syn-1,2-amino alcohols are obtained highly stereoselectively after oxidative cleavage.
25
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
silica gel
sodium benzophenone ketyl
2,3-syn-2-Methoxymethoxy-1,3-nonanediol
(diethylamino)dimethylsilane (HMe2SiNEt2)
chloroplatinic acid hydrate (H2PtCl6·6H2O)
EDTA·2Na
ethanol (64-17-5)
ethyl acetate (141-78-6)
methanol (67-56-1)
ether,
diethyl ether (60-29-7)
ammonium chloride (12125-02-9)
sodium bicarbonate (144-55-8)
sodium chloride (7647-14-5)
sodium carbonate (497-19-8)
PHOSPHORUS (7723-14-0)
sodium sulfate (7757-82-6)
oxygen (7782-44-7)
nitrogen (7727-37-9)
platinum,
platinum(0) (7440-06-4)
cyclohexane (110-82-7)
carbon (7782-42-5)
potassium hydroxide (1310-58-3)
sodium (13966-32-0)
2-BROMOETHANOL (540-51-2)
hydrogen peroxide,
peroxide (7722-84-1)
thiosulfate
Dimethoxymethane (109-87-5)
magnesium sulfate (7487-88-9)
butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
potassium fluoride (7789-23-3)
hexane (110-54-3)
triethylamine (121-44-8)
ethyl vinyl ether (109-92-2)
argon (7440-37-1)
potassium hydrogen carbonate (298-14-6)
Heptanal (111-71-7)
calcium hydride (7789-78-8)
bromo ether
rhodium (7440-16-6)
sodium thiosulfate pentahydrate
sec-butyllithium (598-30-1)
homoallyl (2154-62-3)
phosphorus pentoxide (1314-56-3)
tert-Butyllithium (594-19-4)
potassium tert-butoxide (865-47-4)
2-Methoxymethoxy-1,3-nonanediol
2-Bromoethyl methoxymethyl ether (112496-94-3)
Methoxymethyl vinyl ether (63975-05-3)
tris[2-(2-methoxyethoxy)ethyl]amine (70384-51-9)
2-Methoxymethoxy-1-nonen-3-ol (114675-31-9)
chlorodimethylsilane (1066-35-9)
1,3-divinyltetramethyldisiloxane (2627-95-4)
dimethoxypropane (4744-10-9)
1,1,3,3-tetramethyldisilazane (15933-59-2)
1,3-Nonanediol, 2-(methoxymethoxy)-, (R,R)-(±)- (114675-32-0)
disodium salt of ethylenediaminetetraacetic acid (139-33-3)
vinylsiloxane
chloroplatinic acid hexahydrate (16941-12-1)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved