Org. Synth. 1999, 76, 252
DOI: 10.15227/orgsyn.076.0252
COPPER-CATALYZED CONJUGATE ADDITION OF FUNCTIONALIZED ORGANOZINC REAGENTS TO α,β-UNSATURATED KETONES: ETHYL 5-(3-OXOCYCLOHEXYL)PENTANOATE
[
Pentanoic acid, 5-(3-oxocyclohexyl)-, ethyl ester
]
Submitted by B. H. Lipshutz
1
, M. R. Wood, and R. Tirado.
Checked by Ying Huang and David J. Hart.
1. Procedure
Ethyl 5-(3-oxocyclohexyl)pentanoate
. A 50-mL, round-bottomed flask equipped with a magnetic stir bar and a septum, is cooled under a stream of argon
(Note 1), and charged with
3.0 g of zinc powder (46 mmol
, (Note 2)), followed by
16 mL of tetrahydrofuran (THF)
(Note 3). To this mixture is added
155 μL of 1,2-dibromoethane (338 mg, 1.8 mmol, (Note 4)), and the stirred slurry is heated to ca. 65°C (Note 5) for 1 min using a heat gun. The flask is then placed in a water bath (ca. 23°C) with continued magnetic stirring. After one minute,
155 μL of chlorotrimethylsilane (TMS-Cl) (133 mg, 1.2 mmol, (Note 6)) is added, and the slurry is stirred for 15 min (Note 7). The flask is wrapped in aluminum foil and
6.7 mL of neat ethyl 5-iodovalerate (10.24 g, 40.0 mmol, (Note 8)) is slowly added dropwise over 10 min. The flask is placed in a 35°C oil bath and stirred overnight. Initially, the reaction is very exothermic, and THF can be seen slowly refluxing around the neck of the round-bottomed flask. Dry Ice is used to cool the neck of the flask to avoid leaching the septum with THF. Twelve hours later (overnight, (Note 9)), when the reaction is judged complete by TLC (Note 10), the aluminum foil and the oil bath are removed. The presence of a white "dust" that never quite settles is noted. The reaction is allowed to cool to room temperature and
20 mL of THF
is added to the stirred slurry. Stirring is stopped, the solids are allowed to settle as much as possible (vida supra), and the THF solution of the alkylzinc iodide is transferred via canula to a clean, dry 250-mL, round-bottomed flask equipped with a stir bar and a septum. Two additional washes of the remaining excess zinc with THF (29 mL each) are carried out to ensure complete transfer and proper dilution of the alkylzinc iodide (0.38 M). The solution is cooled to −78°C, and
methyllithium (MeLi) in ether (26 mL, 36 mmol, (Note 11)) is added dropwise over 15 min.
While the alkylzinc is being formed, a 25-mL, round-bottomed flask, equipped with a stir bar and septum is cooled under argon, and charged with
178.9 mg of copper(I) cyanide (CuCN) (2.0 mmol, (Note 12)), followed by
10 mL of THF
. The mixture is cooled to −78°C and
MeLi in ether (2.86 mL, 4.0 mmol, (Note 11)) is added slowly over 10 min. When the addition of MeLi is complete, the slurry is gently warmed until the reaction mixture becomes homogeneous at which point it is recooled to −78°C. With both solutions at −78°C, the higher-order dimethylcyanocuprate is transferred via canula to the alkylzinc reagent. After 5 min at −78°C, 7.77 mL of neat
TMS-Cl (6.65 g, 61.3 mmol, (Note 6)) is added dropwise over 10 min, followed by 10 min of stirring. Finally,
2.02 mL of neat 2-cyclohexen-1-one (2.01 g, 20.9 mmol, (Note 13)) is added dropwise over 15 min via syringe. After 4.5 hr, the reaction is complete as determined by TLC (Note 14) and it is quenched by pouring the reaction mixture into 200 mL of pH 7 buffer (Note 15) and
200 mL of diethyl ether (Et2O) in a 1-L separatory funnel. An additional
25 mL of ether
is used to complete the transfer of the reaction mixture to the separatory funnel. The aqueous layer is further extracted with an additional
50 mL of ether
. The combined organic layers are shaken for 5 min with
50 mL of 1 M tetrabutylammonium fluoride (TBAF) in THF
(Note 16), followed by three washes with
100 mL of brine
. The combined organic phases are dried over anhydrous
magnesium sulfate (MgSO4). Gravity filtration, concentration under reduced pressure, and flash chromatography on
silica gel
(Note 17) using 10:1 (
petroleum ether : ethyl acetate
) affords 4.02 g of slightly contaminated product (Note 18). Warming this material at 50°C on a Kugelrohr apparatus for 2 hr at 0.5 mm affords 3.46 g (73%) of pure 5-(3-oxocyclohexyl)pentanoate as a pale-yellow liquid (Note 19).
2. Notes
1.
All reactions are carried out under an inert atmosphere of
argon (Linde prepurified grade) using oven-dried glassware, syringes, needles and canulas (at least 8 hr, in an
oven at 120°C), employing standard syringe/septa techniques. After dry solids are added to reaction flasks by briefly removing the septum,
argon is passed through the flask to purge it of atmospheric gases for ca. 15 min.
2.
Zinc, powder, −100 mesh, 99.998%, was purchased from the Aldrich Chemical Company, Inc.
, and quickly weighed out on a
bench-top balance.
3.
Tetrahydrofuran is freshly distilled from
sodium benzophenone ketyl
under a
nitrogen atmosphere.
4.
1,2-Dibromoethane, 99+%, purchased from the Aldrich Chemical Company, Inc.
, was used as received.
5.
Gentle refluxing is seen on the sides of the round-bottomed flask, at which time the hot air of the heat gun is directed away from the flask, until the refluxing stops.
6.
Chlorotrimethylsilane, redistilled, 99+%, was purchased from the Aldrich Chemical Company, Inc.
, distilled from
calcium hydride (CaH2) under
argon, and stored under a
Teflon-taped polyethylene cap.
7.
Gas evolution is occasionally observed at this point.
8.
Ethyl 5-iodovalerate is prepared from commercially available
ethyl 5-bromovalerate (Aldrich Chemical Company, Inc.) through a Finkelstein reaction in
acetone
with
sodium iodide
. To
12.5 g (59.5 mmol) of ethyl 5-bromovalerate in 150 mL of dry acetone
is added a total of
44.8 g (299 mmol) of solid sodium iodide
in three equal portions. The solution is warmed under reflux (oil bath temperature of 67°C) for 40 hr and then cooled to room temperature. The mixture is partitioned between
200 mL of diethyl ether
and 200 mL of water. The aqueous phase is extracted with three
100-mL portions of ether
. The combined organic phases are washed with
50 mL of 10% aqueous sodium bisulfite
,
50 mL of brine
, dried (MgSO
4), and concentrated under reduced pressure. The residual oil is distilled (
170-180°C/0.5 mm) to give
14.3 g (
94%) of the iodide:
1H NMR (CDCl
3) δ: 1.24 (t, 3 H, J = 7), 1.72 (m, 2 H), 1.85 (m, 2 H), 2.31 (t, 2 H, J = 7), 3.17 (t, 2 H, J = 7), 4.12 (q, 2 H, J = 7)
. The product is stored over freshly cut pieces of
copper wire
to ensure dryness and long-term purity.
9.
The reaction time for complete
zinc insertion is significantly less than 12 hr (usually 1-4 hr), but additional time at 35°C did not diminish the quality of the
zinc reagent (unless the
zinc reagent contained an enolizable ketone group). An overnight reaction time, for the
zinc oxidative insertion, was used solely for convenience.
10.
The starting iodide is no longer seen by TLC under UV-light, R
f = 0.70 in 10:1 petroleum ether :
ethyl acetate on pre-coated
silica gel 60 F
254 plates (EMx Science), 0.25-mm layer thickness. Further evidence of complete
zinc insertion is seen in the clumping of the excess
zinc into small, shiny metallic balls.
11.
MeLi in ether (1.4 M, low halide) was purchased from the Aldrich Chemical Company, Inc.
and titered against distilled
2-pentanol
, with
1,10-phenanthroline
as indicator.
12.
Copper(I) cyanide, 99%, purchased from the Aldrich Chemical Company, Inc.
, was used as received and stored in an
Abderhalden desiccator over
potassium hydroxide
. It is quickly weighed out on a bench-top balance.
13.
2-Cyclohexen-1-one, 95+%, was purchased from the Aldrich Chemical Company, Inc.
and distilled prior to use.
14.
Only a trace of
2-cyclohexen-1-one, (R
f = 0.16 in 10:1
petroleum ether :
ethyl acetate) could be seen (see
(Note 10) for type of TLC plates used). A new spot at R
f = 0.50 appeared corresponding to the TMS enol ether product if the TLC plate was eluted immediately after spotting. If, however, the TLC plate was not eluted for minutes after spotting, significant cleavage of the TMS enol ether occurred on the
silica gel, allowing observation of the final ketone product at R
f = 0.11.
15.
Aqueous pH 7 buffer was purchased from Fisher Scientific Company. An acidic workup should be avoided so as to prevent the formation of HCN.
16.
1 M TBAF in THF (5% water) was purchased from the Aldrich Chemical Company, Inc.
, and used as received.
17.
Flash chromatography was performed on ICN BioMedical's, ICN Silica, 32-63, 60 Å, using ca.
150 g of silica
in a
2-in diameter column.
18.
Impurities consisted of a small amount of
2-cyclohexen-1-one and another impurity that displayed a triplet at δ 0.9 in its NMR spectrum. These volatile impurities could be removed by warming the crude product under reduced pressure for several hours.
19.
The spectral data are as follows:
1H NMR δ:1.22 (t, 3 H, J = 7, CH
3), 1.31 (m, 5 H), 1.54-2.04 (m, 6 H), 2.19-2.40 (m, 4 H), 2.24 (t, 2 H, J = 7, CH
2CO
2), 4.07 (q, 2 H, J = 7, OCH
2CH
3)
;
13C NMR δ: 14.1 (q), 24.8 (t), 26.0 (t), 26.1 (t), 31.1 (t), 34.1 (t), 36.1 (t), 38.3 (d), 41.3 (t), 47.9 (t), 60.1 (t), 173.4 (s), 211.6 (s)
; IR (neat) cm
−1: 2933, 2861, 1734, 1188
; MS (El), m/e (rel. intensity) 226 (M
+, 2), 181 (7), 135 (5), 101 (7), 98 (8), 97 (100), 82 (6), 81 (7), 67 (7), 55 (16)
; HRMS (El) calcd for [M
+, C
13H
22O
3] 226.1563; found: 226.1569
.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
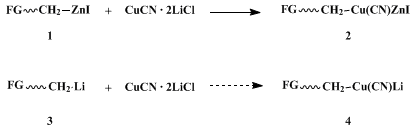
Organocopper chemistry
2 has steadily evolved so that it now includes the preparation and coupling reactions of cuprates bearing ligands that contain electrophilic centers.
3,4,5 Functional groups (FG) such as esters, ketones, nitriles, and halides can be incorporated into lower order cyanocuprates,
2, via metathesis between precursor organozinc halides
1 and CuCN.
3 Use of
zinc in this scheme allows for generation of organometallics such as
1 that would otherwise be difficult to prepare in the corresponding lithiated form (
3) en route to lithiocyanocuprates
4.
6 The reactivity patterns of cuprates
2 reflect the importance of the gegenion in Michael-type additions, for while their lithio counterparts
4 react with most enones quickly at low temperatures,
2 zinc halide cuprates are relatively sluggish.
7 Most significantly, greater than stoichiometric amounts of
copper are normally required. The procedure described here
8 addresses these shortcomings, as well as those of related procedures which, e.g., rely on excesses of
hexamethyl phosphoramide (HMPA).
9
The key feature that allows the cuprate-catalyzed procedure described is the facility with which ligands on
zinc and
copper undergo exchange.
8 Thus, when zinc halide
1 is converted to the mixed
zinc species
5 and then exposed to 5 mol%
Me2Cu(CN)Li2
at low temperatures, catalytic amounts of FG ~CH
2Cu(CN)Li (
4) are produced via transmetallation (along with
Me3ZnLi).
10 In the presence of
TMS-Cl as an activating additive,
11
4 delivers the functionalized ligand to the β-position of an enone (Scheme 1). The intermediate enolate is trapped by the
Me3SiCl present, the Si-O bond being cleaved on workup with fluoride ion to arrive at the product ketone. As anticipated from earlier studies,
12
4 is the most reactive among several species in solution.
10 The overall process is made catalytic in
Cu(I) by virtue of formation of a silyl enol ether, thereby releasing the metal for recycling.
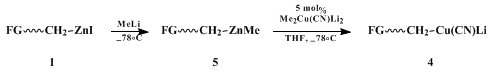
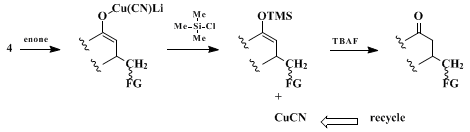
Table I
8 highlights several other examples that demonstrate the scope of the coupling process. Cases of simple ethyl esters (entries 1-3), pivaloates (entries 4,5), chlorides (entries 6,7), a nitrile (entry 8), and a ketone (entry 9), all participate readily. Note the transfer of an acyl silane
13 (entry 10), and the fact that these couplings are effected under very mild conditions and afford good isolated yields.
Table I. Cuprate-Catalyzed 1,4-Additions of Organozinc Reagents8
|
|
Entry
|
Iodide
|
Enone
|
Functionalized Product
|
Yield (%)
|
|
|
1
|
I-(CH2)4CI
|
|
|
89
|
|
2
|
I-(CH2)4CI
|
|
|
83
|
|
3
|
I-(CH2)5CN
|
|
|
85
|
|
4
|
"
|
|
|
85
|
|
5
|
|
|
|
74
|
|
6
|
|
|
|
81
|
|
7
|
|
|
|
85
|
|
8
|
|
|
|
72
|
|
9
|
|
|
|
83
|
|
10
|
|
|
|
74
|
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Zinc (8,9); (7440-66-6)
1,2-Dibromoethane:
Ethane, 1,2-dibromo- (8,9); (106-93-4)
Chlorotrimethylsilane:
Silane, chlorotrimethyl- (8,9); (75-77-4)
Ethyl 5-iodovalerate:
Pentanoic acid, 5-iodo-, ethyl ester (9); (41302-32-3)
Methyllithium:
Lithium, methyl- (8,9); (917-54-4)
Copper(I) cyanide:
Copper cyanide (8,9); (544-92-3)
2-Cyclohexen-1-one HIGHLY TOXIC: (8,9); (930-68-7)
Tetrabutylammonium fluoride:
Ammonium, tetrabutyl-, fluoride (8);
1-Butanaminium, N,N,N-tributyl-, fluoride (9); (429-41-4)
Ethyl 5-bromovalerate:
Valeric acid, 5-bromo-, ethyl ester (8);
Pentanoic acid,
5-bromo-, ethyl ester (9); (14660-52-7)
Sodium iodide (8,9); (7681-82-5)
2-Pentanol (8,9); (6032-29-7)
1,10-Phenanthroline (8,9); (66-71-7)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved