Org. Synth. 2002, 78, 1
DOI: 10.15227/orgsyn.078.0001
(R)-2-DIPHENYLPHOSPHINO-2'-METHOXY-1,1'-BINAPHTHYL
[
Phosphine, (2'-methoxy[1,1'-binaphthalen]-2-yl)diphenyl-,
(R)-
]
Submitted by Yasuhiro Uozumi, Motoi Kawatsura, and Tamio Hayashi
1
.
Checked by Sarge Salman and Louis S. Hegedus.
1. Procedure
Caution! All reactions should be conducted in a well-ventilated hood.
A.
(R)-2,2'-Bis(trifluoromethanesulfonyloxy)-1,1'-binaphthyl
(2).
A dry, 200-mL, two-necked,
round-bottomed flask
is fitted with a magnetic stirring bar and a 30-mL
pressure-equalizing addition
funnel, and flushed with nitrogen gas. The flask is charged with
14.3 g
(50.0 mmol) of (R)-(+)-1,1'-bi-2-naphthol
(1) (Note 1),
12.0 mL
(148 mmol) of pyridine
(Note 2), and
100 mL
of dichloromethane
(Note 3), and the entire mixture is cooled to 0°C with an
ice-water bath.
Trifluoromethanesulfonic anhydride,
(20.0 mL, 33.5 g,
119 mmol)
(Note 2) is added dropwise
over a period of 10 min to the stirred solution.
After the mixture is stirred at 0°C for 6 hr, the reaction mixture is concentrated
on a rotary evaporator.
The residual brown oil is diluted with
200 mL
of ethyl acetate
and transferred to a 500-mL separatory funnel. The organic
phase is washed with
5%
hydrochloric acid
(70 mL),
saturated
sodium bicarbonate (70
mL), and saturated
sodium chloride (70 mL).
The organic phase is dried
over anhydrous sodium sulfate
,
and concentrated under reduced pressure
on a rotary evaporator. The residue is chromatographed (10
× 20 cm column) on silica gel (700 g)
(Note 4). The column is eluted with dichloromethane
and the fractions are analyzed
by TLC on silica gel (Note 5) using
30%
dichloromethane-hexane
as eluant.
Fractions containing the product are
combined and the solvent is evaporated on a rotary evaporator
to give
26.3 g (96%) of
2 as a white powder (Note 6).
B.
(R)-(+)-2-Diphenylphosphinyl-2'-trifluoromethanesulfonyloxy-1,1'-binaphthyl
(3).
A dry, 500-mL, Schlenk tube is fitted with a magnetic
stirring bar
and a rubber septum, and flushed with nitrogen
gas. The flask is charged with
25.0 g (45.4 mmol) of (R)-2,2'-bis(trifluoromethanesulfonyloxy)-1,1'-binaphthyl
(2),
18.4 g (91.0 mmol) of diphenylphosphine
oxide
(Note 7),
1.02 g (4.54 mmol) of palladium
acetate
(Note 8),
1.94 g (4.55
mmol) of
1,4-bis(diphenylphosphino)butane (dppb)
(Note 9),
23.4 g of diisopropylethylamine
(181 mmol)
(Note 10) and
200 mL
of dimethyl sulfoxide
(Note 11), and the entire mixture is heated with stirring at
100°C for 12 hr (Note 12).
After the reaction mixture is cooled to room temperature, it is concentrated under
reduced pressure (0.1-0.2 mm) on a rotary
evaporator. The dark brown residue is diluted with
400
mL of ethyl
acetate
and transferred to a 1-L separatory funnel.
The organic phase is washed successively
with water (two 100-mL portions),
5%
hydrochloric
acid (100
mL), saturated
sodium bicarbonate
(100 mL),
and saturated
sodium chloride (100
mL), and the organic phase
is dried over anhydrous sodium sulfate
.
After filtration the organic phase is
concentrated under reduced pressure on a rotary evaporator,
and the residue is chromatographed (14 × 30-cm
column) on silica gel (ca. 1.5 kg) (Note 4). The column is eluted
with
50%
ethyl acetate-hexane
and the
fractions are analyzed by TLC on silica gel (Note 5) using the same eluant. Fractions containing the product are combined
and the solvent is evaporated on a
rotary evaporator to give 23.8
g (87%) of 3 as a white powder (Notes
13,
14).
C.
(R)-(−)-2-Diphenylphosphinyl-2'-hydroxy-1,1'-binaphthyl
(4)
. A 100-mL round-bottomed flask
containing a magnetic stirring bar is charged with
6.07 g (10.1 mmol) of (R)-(+)-2-diphenylphosphinyl-2'-trifluoromethanesulfonyloxy-1,1'-binaphthyl
(3),
30 mL of 1,4-dioxane
,
and
14 mL of methanol
.
(Note 15) To the solution is added
14.1
mL of 3 N aqueous sodium hydroxide (NaOH)
solution at ambient temperature. The reaction mixture is stirred for 12 hr then acidified
(to pH 1) by the addition of a few drops of concd hydrochloric acid.
The mixture is transferred to a separatory funnel and extracted twice with ethyl
acetate (EtOAc). The organic phase is dried over
magnesium
sulfate
(MgSO4), filtered, and concentrated under
reduced pressure to afford 6.19 g
of 4 as a solid. (Note 16). This crude material is carried
on to the next step without purification, assuming 100% yield.
D.
(R)-(+)-2-Diphenylphosphinyl-2'-methoxy-1,1'-binaphthyl
(5).
A 250-mL round-bottomed flask is charged with crude
4, 5.55 g (40.2 mmol) of potassium
carbonate(K2CO3), and
66
mL of acetone
. To this mixture is added
2.5 mL (40.2 mmol) of methyl
iodide (MeI)
(Note 17). The reaction mixture
is refluxed for 3 hr. After the reaction is cooled to room temperature it is filtered
through a Celite pad (Note 18), and the filter cake is washed
with
diethyl ether (Et2O).
The filtrate is concentrated under reduced pressure to give 6.88 g of 5 as a brown powder (Note 19).
This crude material is carried on to the next step without purification, assuming
100% yield.
E.
(R)-(+)-2-Diphenylphosphino-2'-methoxy-1,1'-binaphthyl
(6).
A 250-mL round-bottomed flask containing a magnetic stirring
bar is charged with crude 5,
7 mL
(50 mmol) of triethylamine (Et3N)
(Note 20), and
84 mL
of toluene
. (Note 21). The mixture
is cooled to 0°C then
4 mL (40
mmol) of trichlorosilane (Cl3SiH)
(Note 22)
is added via syringe. The reaction is heated to 120°C and stirred for 5 hr. Upon
cooling to ambient temperature the reaction is diluted with Et2O and quenched
with aqueous saturated sodium bicarbonate. The resulting suspension
is filtered through a Celite pad, and the filter cake is washed with Et2O.
The organic phase is dried over MgSO4 then concentrated under reduced pressure
to afford 4.94 g of a yellow solid. This crude material is dissolved in a minimal
amount of dichloromethane (CH2Cl2) and
chromatographed on a 10 × 30-cm column containing 615 g of silica gel (SiO2).
The column is eluted with Et2O to afford 4.08
g (8.71 mmol, 86% yield over last 3 steps) of 6
as an off-white powder (Note 23).
2. Notes
1.
(R)-(+)-1,1'-Bi-2-naphthol
(>99% op) was purchased from Aldrich Chemical Company, Inc.
,
and used without further purification.
2.
Pyridine
and
trifluoromethanesulfonic anhydride
were purchased from Aldrich Chemical Company, Inc.
,
and used without further purification.
3.
Dichloromethane
was purchased from Fisher Scientific, and distilled from calcium hydride
before use.
4.
Silica gel 60 230-400 mesh ASTM was used.
5.
Merck silica gel 60F-254 plates were used.
6.
Specific rotation value of
2:
[α]D
22 −143.5° (CHCl3,
c 1.24) [literature rotation for (S)-
2;
2
[α]D
22 +142°
(CHCl3, c 1.035)];
1H
NMR δ: 7.27 (d, 2 H, J = 8.7), 7.41 (t, 2 H, J = 7.8),
7.58 (t, 2 H, J = 7.8), 7.62 (d, 2 H, J = 9.6), 8.00
(d, 2 H, J = 8.3), 8.13 (d, 2 H, J = 9.2).
If the material does not solidify, addition of equal amounts of
diethyl
ether and
hexane, followed by reevaporation, should
produce solid material.
7.
Diphenylphosphine oxide
is commercially available from Aldrich Chemical Company, Inc.
,
and was recrystallized from
1:1 hexanes/ethyl acetate
prior to use. The submitters have prepared and used this reagent. Preparation of
diphenylphosphine oxide
: To a solution
of
diethyl phosphite (65.0
mL, 505 mmol) in
250
mL of tetrahydrofuran
(THF) is added
sodium metal (11.5 g, 500
mg-atom) and the mixture is stirred under reflux for 20 hr. The
resulting solution is added to
1.9 M solution (THF/Et2O
= 1/2) of phenylmagnesium bromide (580 mL, 1.10
mol) at 0°C and the mixture is refluxed for 6 hr. After the
mixture is quenched with a small amount of water, it is diluted with
ethyl
acetate and washed once with
5%
hydrochloric acid (HCl) and twice with water. The
organic phase is dried over
anhydrous sodium sulfate
and concentrated under reduced pressure to give crude
diphenylphosphine
oxide
. The crude solid is purified by silica gel column chromatography
(eluant:
ethyl acetate) to give
diphenylphosphine
oxide
(77.0 g, 75%).
8.
Palladium acetate
was purchased from Aldrich Chemical Company, Inc.
,
and purified as follows:
Palladium acetate
is dissolved in hot
benzene
and filtered from insoluble material. After removal of the solvent, the residue is
triturated with a small amount of diethyl ether to give brown powder that is collected
by filtration, washed with diethyl ether and dried.
9.
1,4-Bis(diphenylphosphino)butane
was purchased from Aldrich Chemical Company, Inc.
,
and used without further purification.
10.
Diisopropylethylamine
was purchased from Aldrich Chemical Company, Inc.
,
and used without further purification.
11.
Dimethyl sulfoxide
was purchased from Aldrich Chemical Company, Inc.
,
and distilled from calcium hydride before use.
12.
The use of
5 mol% of
the catalyst [Pd(OAc)2-dppb] also gave a high yield of
3,
but the submitters recommend the use of 10 mol % of the catalyst to ensure high chemical
yield in the 12-hr reaction.
13.
The physical properties of
3 are as follows:
[α]D
20 +44.4° (CHCl3,
c 1.20),
[α]D
22
+7.4° (CH2Cl2, c 1.40) [literature rotation
for (R)-
3;
3
[α]D +6.29° (CH2Cl2,
c 1.00)]; IR (KBr) cm
−1
ν: 1410, 1205, 1140, and 945
;
1H NMR δ: 6.9-8.1 (m, 22 H, aromatic)
;
31P NMR δ: 28.9 (s)
;
EIMS m/z 603 (M+1), 454, 201 (base peak)
.
Anal. Calcd for C
33H
22F
3O
4PS: C, 65.78;
H, 3.68. Found: C, 65.67; H, 3.89.
14.
Assignment of all peaks in the
13C NMR is difficult
because of
13C-
31P coupling and the overlapping of peaks.
15.
Methanol and 1,4-dioxane
were purchased from Aldrich Chemical Company, Inc.
,
and used without further purification.
16.
An
analytically pure sample is isolated
by column chromatography on silica gel (see Note
4). The column is eluted with
50%
ethyl acetate-hexane
and the
fractions are analyzed by TLC on silica gel (see Note
5) using the same eluent. Fractions containing the product are combined
and the solvent is evaporated on a
rotary evaporator to give
4 as a white powder. The physical properties of
4 are as follows (see
Note
14):
[α]D
20
−105° (CHCl3, c 0.55),
[α]D
20 −113° (CH2Cl2,
c 1.00) [literature rotation for (R)-
4;
3
[α]D −108.3° (CH2Cl2,
c 1.00)];
1H
NMR δ: 6.35-8.10 (m, 22 H), 9.01 (br s, 1 H)
;
31P NMR δ: 31.42 (s)
;
EIMS m/z 470 (M
+), 268 (base
peak)
; HRMS calcd for C
32H
23PO
2
470.1436, found 470. 1415. Anal. Calcd for C
32H
23O
2P:
C, 81.69; H, 4.93. Found: C, 81.66; H, 4.96.
17.
Methyl iodide, potassium
carbonate, and acetone were purchased from Aldrich
Chemical Company, Inc.
, and used without further purification.
18.
Celite 535 (45 gals/sq.ft/hour), purchased
from J.T. Baker, was used.
19.
An analytically pure sample is isolated by column chromatography
on silica gel (see Note
4). The column is
eluted with
ethyl acetate and the fractions are analyzed by TLC
on silica gel (see Note
5) using the same
eluent. Fractions containing the product are combined and the solvent is evaporated
on a
rotary evaporator to give
5 as a white powder.
The physical properties of
5 are as follows
(Note 14):
[α]D
20 +121.5°
(CHCl3, c 1.30);
1H
NMR δ: 3.58 (s, 3 H), 6.75-8.01 (m, 22 H);
31
P NMR δ: 28.88(s)
;EIMS m/z 484 (M
+), 453, 282 (base peak)
;
HRMS calcd for C
33H
25O
2P 484.1592, found 484.1574.
Anal. Calcd for C
33H
25O
2P: C, 81.80; H, 5.20. Found:
C, 81.77; H, 5.38.
20.
Triethylamine was
purchased from Aldrich Chemical Company, Inc., and used without
further purification.
21.
Toluene was purchased
from Aldrich Chemical Company, Inc., and distilled from calcium
hydride before use.
22.
Trichlorosilane
was purchased from Aldrich Chemical Company, Inc.
,
and used without further purification. While it is difficult to measure the volume
of
trichlorosilane used accurately because of its volatility,
accurate measurement is not essential in the reduction of
5. The submitters
found that a large excess of
trichlorosilane does not interfere
with reduction of phosphine oxide in Part E, and recommend the use of trichlorosilane
(4 equiv or more to
5) to complete the reduction in an appropriate reaction
time.
23.
Crystallization of the crude material from
dichloromethane-hexane
gave product
6 of 50% yield or lower. For efficiency of isolation, the submitters
recommend purifying
6 by column chromatography. The physical properties of
product
6 are as follows (see Note
14):
[α]D
20 +95°
(CHCl3, c 0.27),
[α]D
16
+75.7° (benzene, c 1.50) [literature rotation for (S)-
6;
4
[α]D
16−59.3°
(benzene, c 1.0)];
mp 174-176°C
(recrystallization from CH
2Cl
2/n-hexane);
1H NMR δ: 3.34 (s, 3 H), 6.95-8.05 (m, 22 H)
;
31P NMR δ: −12.74
(s)
; EIMS m/z 468
(M
+), 437 (base peak)
; HRMS calcd for C
33H
25PO
2
468.1643, found 468.1672. Anal. Calcd for C
33H
25OP: C, 84.60;
H, 5.38. Found: C, 84.35; H, 5.44.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
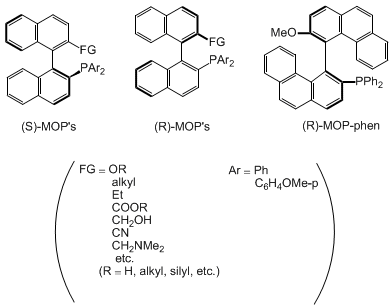
3. Discussion
Most of the chiral phosphine ligands prepared so far and used for catalytic
asymmetric reactions are the bisphosphines,
5 which are expected to construct an effective
chiral environment by bidentate coordination to metal; they have been demonstrated
to be effective for several types of asymmetric reactions. On the other hand, there
exist transition metal-catalyzed reactions where the bisphosphine-metal complexes
cannot be used because of their low catalytic activity and/or low selectivity towards
a desired reaction pathway. Therefore chiral monodentate phosphine ligands
are required for the realization of new types of catalytic asymmetric reactions. Unfortunately,
only a limited number of monodentate chiral phosphine ligands have been reported,
6
which with few exceptions are not so useful as bisphosphine ligands. Recently, the
monodentate, optically active phosphine ligand,
2-diphenylphosphino-2'-methoxy-1,1'-binaphthyl
(MeO-MOP), and its analogs
7
have been demonstrated to provide high enantioselectivity in palladium-catalyzed hydrosilylation
of olefins
8 and
palladium-catalyzed reduction of allylic esters by
formic acid.
9 The
procedures described here allow the convenient preparation of MOP and have advantages
over previously published sequences.
4 MeO-MOP can be
prepared in five steps from binaphthol without racemization and the overall yield
is 72%. The key step in this process is the palladium-catalyzed monophosphinylation
of
2,2'-bis(trifluoromethanesulfonyloxy)-1,1'-binaphthyl
,
which was originally reported by Morgans and co-workers.
3
Under the slightly modified conditions, ditriflate (R)-
2 was efficiently converted
into (R)-
3 (87% yield) without racemization. Hydrolysis of the remaining triflate
with aqueous
sodium hydroxide
in
1,4-dioxane
and
methanol
(2/1) gave (R)-
4. The
phenolic hydroxyl group of (R)-
4 was methylated by treatment with
methyl
iodide in the presence of
potassium
carbonate
in acetone to give (R)-
5. Reduction of
phosphine oxide
(R)-
5 was carried
out with trichlorosilane and triethylamine
10
and references cited therein. in toluene with heating to give the corresponding phosphine
(R)-
6 (86% yield over last 3 steps).
A variety of MOP derivatives bearing various alkoxy or siloxy groups were readily
prepared by changing the reagent used for the alkylation of
4.
7 Furthermore, the presence of the triflate group
11 in compound
3 allows one to prepare a wide range
of MOP derivatives functionalized at the 2'-position. Thus, the 2'-alkyl,
carboxyl, cyano, aminomethyl groups, etc. were introduced into the 2'-position
of MOP via transition metal-catalyzed cross-coupling, carbonylation, and cyanation
reactions.
7,12 Bis(substituted phenyl)phosphino groups were readily
introduced into the binaphthyl by the palladium-catalyzed reaction with the corresponding
diarylphosphine oxides. The same procedures used for the preparation of
3 were
followed with di(p-methoxyphenyl)phosphine oxide and
2. Subsequent hydrolysis,
alkylation, and reduction processes gave 2-di(p-methoxyphenyl)phosphino-MOP.
7 The flexibility of the synthetic route allows fine tuning of the
phosphine ligand by the introduction of several types of side chains and control of
the steric and electronic effects of the phosphino group. Needless to say, the synthetic
procedures shown here can be used for the preparation of MOPs having the (S)-absolute
configuration by using (S)-binaphthol as a starting material. In addition, a MOP analog
having the biphenanthryl skeleton (MOP-phen) was also prepared from optically active
4,4'-biphenanthrol through the same sequences mentioned above.
9b
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(R)-2-Diphenylphosphino-2'-methoxy-1,1'-binaphthyl:
Phosphine, (2'-methoxy[1,1'- binaphthalen]-2-yl)diphenyl-, (R)-
(12);(145964-33-6)
(R)-2,2'-Bis(trifluoromethanesulfonyloxy)-1,1'-binaphthyl:
Methanesulfonic acid, trifluoro-, [1,1'-binaphthalene]-2,2'-diyl
ester, (R)- (12); (126613-06-7)
(R)-(+)-1,1'-Bi-2-naphthol:
[1,1'-Binaphthalene]-2,2'-diol,
(R)-(+)- (8);
[1,1'-Binaphthalene]-2,2'-diol, (R)-
(9); (18531-94-7)
Pyridine (8,9); (110-86-1)
Trifluoromethanesulfonic anhydride:
Methanesulfonic
acid,
trifluoro-, anhydride (8,9); (358-23-6)
(R)-(+)-2-Diphenylphosphinyl-2'-trifluoromethanesulfonyloxy-1,1'-binaphthyl:
Methanesulfonic acid, trifluoro-, 2'-(diphenylphosphinyl)[1,1'-binaphthalen]-2-yl
ester, (R)- (12); (132532-04-8)
Diphenylphosphine oxide:
Phosphine oxide,
diphenyl- (8,9); (4559-70-0)
Palladium acetate:
Acetic acid, palladium(2+)
salt(8,9); (3375-31-3)
1,4-Bis(diphenylphosphino)butane (dppb):
Phosphine,
tetramethylenebis[diphenyl- (8);
Phosphine, 1,4-butanediylbis[diphenyl-(9);
(7688-25-7)
N,N-Diisopropylethylamine:
Triethylamine,
1,1'-dimethyl- (8);
2-Propanamine, N-ethyl-N-(1-methylethyl)-
(9); (7087-68-5)
Dimethyl sufoxide:
Methyl sulfoxide
(8);
Methane, sulfinyl bis- (9); (67-68-5)
(R)-(−)-2-Diphenylphosphinyl-2'-hydroxy-1,1'-binaphthyl:
[1,1'-Binaphthalene]-2-ol, 2'-(diphenylphosphinyl)-, (R)-
(12); (132548-91-5)
1,4-Dioxane: CANCER SUSPECT AGENT:
p-Dioxane
(8);
1,4-Dioxane (9); (123-91-1)
(R)-(+)-2-Diphenylphosphinyl-2'-methoxy-1,1'-binaphthyl:
Phosphine oxide, (2'-methoxy[1,1'-binaphthalen]-2-y1)diphenyl-,
(R)- (13); (172897-73-3)
Methyl iodide:
Methane, iodo-(8,9);
(74-88-4)
Triethylamine (8);
Ethanamine, N,N-diethyl-(9);
(121-44-8)
Trichlorosilane:
Silane, trichloro-
(8,9); (10025-78-2)
Diethyl phosphite:
Phosphonic acid, diethyl
ester (8,9); (762-04-9)
Sodium (8,9); (7440-23-5)
Phenylmagnesium bromide:
Magnesium, bromophenyl-
(8,9); (100-58-3)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved