Org. Synth. 2002, 78, 63
DOI: 10.15227/orgsyn.078.0063
ULLMAN METHOXYLATION IN THE PRESENCE OF A 2,5-DIMETHYLPYRROLE-BLOCKED
ANILINE: PREPARATION OF 2-FLUORO-4-METHOXYANILINE
[
Benzenamine, 2-fluoro-4-methoxy-
]
Submitted by John A. Ragan, Brian P. Jones, Michael J. Castaldi, Paul D. Hill, and Teresa W. Makowski
1
.
Checked by Samuel W. Ridenour and David J. Hart.
1. Procedure
A.
1-(2-Fluoro-4-iodophenyl)-2,5-dimethyl-1H-pyrrole
.
A 500-mL, single-necked, round-bottomed flask with a Teflon-coated
magnetic stir bar is charged with
2-fluoro-4-iodoaniline
(50.0 g, 211 mmol)
(Note 1),
p-toluenesulfonic acid (0.40 g,
2.1 mmol)
(Note 2),
250
mL of toluene
(Note 3),
and
acetonylacetone(29.7 mL,
253 mmol)
(Note 4). A Dean-Stark
trap is attached to the flask, and the solution is warmed to reflux for
1 hr (Note 5). After the solution is cooled to room temperature,
it is transferred to a 500-mL separatory funnel, and washed
with one 50-mL portion of aqueous saturated sodium
bicarbonate
(NaHCO3), five 50-mL portions of water
(Note 6), and one 50-mL portion of brine.
The organic phase is dried over anhydrous magnesium
sulfate
(MgSO4), filtered, and concentrated to provide
a brown, free-flowing solid (67.5 g,
101.5% of theory) (Notes 7 and 8).
B.
1-(2-Fluoro-4-methoxyphenyl)-2,5-dimethyl-1H-pyrrole
.
A 500-mL, single-necked, round-bottomed flask equipped with
a condenser and Teflon-coated magnetic stir bar
is flame-dried under a positive pressure of nitrogen, and charged
with
1-(2-fluoro-4-iodophenyl)-2,5-dimethyl-1H-pyrrole
(66.6 g, 211 mmol)
,
sodium
methoxide (NaOMe) (34.2 g, 633 mmol)
(Note 9),
copper(I) chloride
(3.13 g, 31.7 mmol)
(Note 10),
230 mL of methanol
(Note 11) and
70 mL
of dimethylformamide
(DMF) (Note 12).
The resulting slurry is placed in an 80°C oil bath for 90 min
(Note 13) so that a gentle reflux is maintained, then cooled
to room temperature. The slurry is poured into a rapidly stirring mixture of
500 mL of isopropyl ether
(Note 14),
220 mL of
aq 5%
ammonium chloride
(NH4Cl)
(Note 15), and 350 mL of water in a 2-L Erlenmeyer
flask, rinsing with several small portions of methanol.
The resulting slurry is stirred overnight (Note 16). It is filtered
through a 3"-pad of Celite in a coarse-frit glass
funnel, and the filtrate is transferred to a 2-L separatory
funnel. The phases are separated, and the aqueous phase is extracted with
three 50-mL portions of isopropyl ether
.
The combined organic phases are washed with 200 mL of aqueous 10% NH4OH
(Note 17), filtered through a silica gel pad (Note 18),
and concentrated to provide a brown, free-flowing solid (42.0 g, 92%
crude yield) (Note 19). The crude product is recrystallized by
dissolving it in 100 mL of hot hexanes and stirring overnight at room temperature,
then collecting the resulting brown solid (34.7
g, 75% yield) (Note 20).
C.
2-Fluoro-4-methoxyaniline
.
A 1-L, single-necked flask equipped with a reflux
condenser and Teflon-coated magnetic stirring bar
is charged with
1-(2-fluoro-4-methoxyphenyl)-2,5-dimethyl-1H-pyrrole
(34.7 g, 158 mmol)
,
hydroxylamine
hydrochloride (110 g, 1.58 mol)
(Note 21),
triethylamine
(44.0 mL, 316 mmol)
(Note 22),
300 mL of 95% ethanol,
and 150 mL of water. The resulting solution is warmed to reflux for 20 hr (Notes 23 and 24),
then cooled to room temperature. The reaction is quenched by pouring into a rapidly
stirred solution of 200-300 mL of ice-cold
1 N hydrochloric acid (HCl). This solution is washed with two 250-mL portions of isopropyl ether
,
the pH is adjusted to 9-10 by careful addition of 6 N sodium
hydroxide
(NaOH), and the resulting mixture is extracted with
two 250-mL portions of isopropyl ether
.
The final organic phase is dried over MgSO4, filtered, and concentrated
to an oily, brown solid. This material is triturated with several portions of isopropyl
ether with warming on a steam bath to dissolve as
much product as possible, decanting away from an insoluble cream colored solid (Note 25). Concentration of these extracts provides a brown solid (18.1 g, 81%
crude yield), which is recrystallized by dissolving in
18
mL of hot isopropyl ether
, and slowly adding
80 mL of hexanes. After the product is placed in an ice bath
for 1 hr, it is isolated as a brown solid (16.0
g, 72% yield), which
is further purified by trituration at room temperature with 160 mL of water for 16
hr (Note 26): filtration and drying in a vacuum oven (with little
if any heating) provide analytically pure product: 13.0
g, 58% yield (Note 27).
2. Notes
1.
2-Fluoro-4-iodoaniline
was obtained from Aldrich Chemical Company, Inc.
,
and used without further purification. It can also be prepared by iodination of
2-fluoroaniline.
2
2.
p-Toluenesulfonic acid monohydrate
was obtained from Aldrich Chemical Company, Inc.
,
and used without further purification.
3.
A.C.S. reagent grade toluene
was obtained from J. T. Baker and used as received.
4.
Acetonylacetone(hexane-2,5-dione)
was obtained from Aldrich Chemical Company, Inc.
,
and used without further purification.
5.
The reaction can be monitored by TLC (
20:1
hexane-ethyl acetate
, UV visualization,
SM R
f = 0.11, product R
f = 0.56). The submitters indicate that
the reaction can also be monitored by GC/MS (Hewlett-Packard 5890 GC/MS, HP-1 column
(12 m×0.2 mm×0.33 µm), 1 mL/min flow rate, injector temp. 280°C, oven temp. 133°C
for 0.1 min, then ramp 19°C/min to 310°C, hold for 1.65 min): SM R
f = 1.53
min, product R
f = 3.10 min.
6.
Multiple aqueous washes assist in removing any excess
acetonylacetone.
7.
1H NMR indicates reasonably pure product, with trace
amounts of
toluene and
acetonylacetone.
8.
The product shows the following physical properties:
mp 68-70°C;
1H NMR (400 MHz, CDCl
3) δ: 2.03
(s, 6 H), 5.94 (s, 2 H), 7.02 (t, 1 H, J = 8), 7.64
(m, 2 H)
;
13C
NMR (CDCl
3) (8 of 9 lines observed) δ: 12.5, 92.8,
106.5, 126.1 (d, J = 23), 128.9, 131.9,
134.0 (d, J = 4), 158.1 (d, J = 254)
; MS (EI): m/z 268 (100)
; HRMS
(FAB) calcd for C
12H
11NFI (M
+) 315.9999, found 315.9995.
9.
Sodium methoxide
was obtained from Aldrich Chemical Company Inc.
, and
used without further purification. Out of a total of five separate batches of NaOMe
(all new, unopened bottles) used over a 24-month period, the submitters had one occasion
where the reaction failed to proceed beyond 15-20% conversion. When a new bottle of
NaOMe (different lot number) was used, the reaction worked as usual. The NaOMe that
failed in the Ullman coupling was found to have limited solubility in
methanol
(MeOH) (bottles that worked displayed MeOH solubilities of >100 mg/mL), suggestive
of contamination by significant quantities of NaOH, possibly from adventitious water
introduced during re-packaging or manufacture.
10.
Cuprous chloride
(CuCl) was obtained from Aldrich Chemical Company, Inc.
,
and used without further purification.
11.
A.C.S. reagent grade methanol
was obtained from J. T. Baker
and used as received.
12.
Sure-seal DMF was obtained from Aldrich
Chemical Company, Inc.
, and used as received.
13.
The submitters indicate that the reaction can be monitored by
GC/MS (same conditions as
(Note 5) ): SM R
f = 3.10
min, product R
f = 2.50 min.
14.
A.C.S. reagent grade isopropyl
ether was obtained from J. T. Baker
and
used as received.
15.
The
ammonium chloride
solution was prepared by dissolution of 50 g of NH4Cl (Aldrich
Chemical Company, Inc.) in 950 mL of distilled water.
16.
The
Erlenmeyer is loosely capped with a piece of aluminum
foil or a cork stopper. The submitters have run this step, which assists
in removal of copper salts, for as short as 16 hr to as long as 60 hr with no change
in outcome.
17.
The
ammonium hydroxide was prepared by dilution
of commercial 28-30% NH
4OH (Baker) with 9 volumes of distilled water.
18.
Silica gel, 100 g of 230-400
mesh, was used in an 8-cm diameter,
medium-frit glass funnel.
19.
The crude product is quite pure by
1H NMR, and can
be carried directly into the deprotection if desired.
20.
The product shows the following physical properties:
mp 67-69°C; H NMR (400 MHz, CDCl
3) δ:1.97 (s, 6 H),
3.82 (s, 3 H), 5.89 (s, 2 H), 6.73 (s, 1 H),
6.75 (d, 1 H, J = 8), 7.12 (t, 1 H, J = 8)
;
13C NMR (CDCl
3) (9 of 10 lines
observed) δ: 12.4, 55.7, 102.3 (d, J = 25),
105.6, 106.0, 109.9 (d, J = 3), 129.5,
130.7, 159.1 (d, J = 260)
; MS (EI): m/z 219 (100); HRMS (FAB) calcd for C
13H
14NFO
(M+H) 220.1138, found 220.1127. Product color ranges from light brown to dark brown
with no effect on physical and spectral properties.
21.
Hydroxylamine hydrochloride
was obtained from Fisher Scientific Company
and used
as received.
22.
A.C.S. reagent grade
triethylamine
was obtained from J. T. Baker
and used as received.
23.
Because of the potentially explosive nature of
hydroxylamine
3 the reaction
should be kept behind a blast shield while heating.
24.
The submitters indicate that the reaction can be monitored by
GC/MS (same conditions as Note 5): SM R
f = 2.50 min, product R
f
= 0.90 min.
25.
1H NMR of this material shows just two singlets (δ
2.48 and 1.91), suggesting that it is derived from
acetonylacetone
(possibly the bis-oxime).
26.
This water reslurry serves to remove trace residues of the δ
2.48 and 1.91 impurity referred to in Note 25.
27.
The product shows the following physical properties (ref
4):
mp 46.8-47.1°C;
1H NMR (300 MHz, CDCl
3) δ: 3.34 (br s, 2 H),
3.72 (s, 3 H), 6.53 (m, 1 H), 6.61 (dd, 1 H, J = 3,
10), 6.72 (dd, 1 H, J = 9, 10)
;
13C NMR (100 MHz, CDCl
3) δ:56.2,
102.7, 110.2, 118.1, 128.1, 153.3,
152.4
. Anal. Calcd for C
7H
8NFO: C, 59.57;
H, 5.71; N, 9.92. Found: C, 59.62; H, 5.74; N, 9.99. The product color ranges from
light brown to dark brown with no effect on physical or spectral properties.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
2-Fluoro-4-methoxyaniline has been previously prepared by
nitration of
2-fluorophenol
5 followed
by alkylation with dimethyl sulfate and reduction of the nitro group.
4 Nitration delivers a mixture of regioisomers that require chromatographic
separation. The submitters recently required a practical, multi-gram synthesis of
this compound, and concerns with the potential thermal hazards of a nitration reaction,
poor nitration regioselectivity, and handling of dimethyl sulfate led them to investigate
an alternative synthesis. The commercial availability of 2-fluoro-4-iodoaniline motivated
them to investigate an Ullman coupling.
6They investigated a variety
of blocking groups for the aniline moiety (the unprotected aniline failed to couple
under standard conditions), and found the 2,5-dimethylpyrrole blocking group to be
uniquely suited for this purpose.
7Several
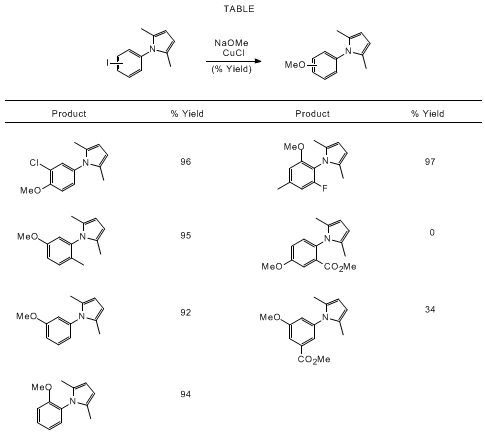
other substrates were also investigated, as summarized in the Table. Interestingly,
electron-withdrawing groups on the aromatic ring led to significantly lower yields
(e.g., entries 7 and 8). However, the regiochemistry between the
2,5-dimethylpyrrole
substituent and the iodide was of little consequence to the yield of methoxylation.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2-Fluoro-4-methoxyaniline (8,9); (458-52-6)
1-(2-Fluoro-4-iodophenyl)-2,5-dimethyl-1H-pyrrole:
1H-Pyrrole,
1-(2-fluoro-4-iodophenyl)-2,5-dimethyl- (14); (217314-30-2)
2-Fluoro-4-iodoaniline:
Aniline, 2-fluoro-4-iodo-
(8,9); (29632-74-4)
p-Toluenesulfonic acid monohydrate (8);
Benzenesulfonic
acid, 4-methyl-, monohydrate (9); (6192-52-5)
Acetonylacetone:
2,5-Hexanedione
(8,9); (110-13-4)
1-(2-Fluoro-4-methoxyphenyl)-2,5-dimethyl-1H-pyrrole:
1H-Pyrrole,
1-(2-fluoro-4-methoxyphenyl)-2,5-dimethyl- (14); (217314-31-3)
Sodium methoxide:
Methanol, sodium salt
(8,9); (124-41-4)
Copper(I) chloride:
Copper chloride
(8,9); (7758-89-6)
N,N-Dimethylformamide:CANCER SUSPECT AGENT:
Formamide,
N,N-dimethyl- (8,9); (68-12-2)
Hydroxylamine hydrochloride (8);
Hydroxylamine,
hydrochloride (9); (5470-11-1)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved