Org. Synth. 2008, 85, 158
DOI: 10.15227/orgsyn.085.0158
SYNTHESIS OF (2R,3R)-2,3-DIMETHYL-1,4-BUTANEDIOL BY OXIDATIVE HOMOCOUPLING OF (4S)-ISOPROPYL-3-PROPIONYL-2-OXAZOLIDINONE
[2,3-dimethylbutane-1,4-diol, (2R, 3R)-]
Submitted by Chong-Dao Lu and Armen Zakarian
1.
Checked by Katrien Brak and Jonathan A. Ellman.
1. Procedure
A. (4S)-Isopropyl-3-propionyl-2-oxazolidinone (2). An oven-dried, 250-mL, three-necked, round-bottomed flask is equipped with a mechanical stirrer, an internal thermometer, and an adapter equipped with an argon inlet and a rubber septum. The flask is flushed with argon and is charged with 333 mL of dry tetrahydrofuran (THF) (Note 1) and (4S)-isopropyl-2-oxazolidinone (8.61 g, 66.7 mmol) (Note 2). The mixture is cooled to −75 °C in a dry ice-acetone bath. At this temperature, a 2.57 M solution of n-butyllithium in hexane (27.2 mL, 70.0 mmol) (Note 3) is added dropwise over 20 min using a syringe at such a rate that the reaction temperature remains below −72 °C. The reaction mixture is stirred at this temperature for 25 min and propionyl chloride (6.29 g, 5.94 mL, 68.0 mmol) is added dropwise over 10 min using a syringe while maintaining an internal temperature below −73 °C. The reaction mixture is stirred at this temperature for 30 min, and the reaction is then quenched with sat. aq. ammonium chloride solution (100 mL). The reaction mixture is transferred to a 1-L separatory funnel and the organic phase is collected. The aqueous phase is extracted with ethyl acetate (3 × 100 mL). The combined organic phases are washed with brine (100 mL). Drying with anhydrous sodium sulfate (40 g), filtration, and removal of the solvent under reduced pressure on a rotary evaporator (30 °C, 40 mmHg) afford the crude product as a light yellow oil. The oil is purified by silica gel column chromatography (Note 4). Fractions containing the product (Note 5) are concentrated by rotary evaporation (30 °C, 40 mmHg) and are dried (Note 6) to yield 11.0 g (89%) of (4S)-isopropyl-3-propionyl-2-oxazolidinone (2) as a colorless oil (Note 7).
B. (2R,3R)-1,4-Bis[(4S)-4-isopropyl-2-oxo-(1,3-oxazolidine-3-yl)]-2,3-dimethylbutane-1,4-dione (3). An oven-dried, 500-mL, three-necked, round-bottomed flask is equipped with an internal thermometer, a 125-mL pressure-equalizing addition funnel fitted with a rubber septum, an adapter equipped with an argon inlet and a rubber septum, and a Teflon-coated magnetic stirring bar (Note 8). The flask is flushed with argon and charged with dry THF (200 mL) (Note 1) and diisopropylamine (6.37 g, 8.83 mL, 63.0 mmol) (Note 9). The mixture is cooled to −75 °C in a dry ice-acetone bath. At this temperature, a 2.52 M solution of butyllithium in hexane (23.8 mL, 60.0 mol) (Note 3) is added dropwise over 10 min using a syringe while maintaining an internal temperature below −70 °C. The reaction mixture is stirred at this temperature for 60 min and a solution of (4S)-isopropyl-3-propionyl-2-oxazolidinone (9.26 g, 50.0 mmol) in THF (50 mL) is added dropwise over 20 min from the dropping funnel, which is rinsed with THF (5 mL), all while maintaining an internal temperature below −71 °C. The reaction mixture is stirred at −75 °C for 45 min. Then, titanium (IV) chloride (23.7 g, 13.7 mL, 125 mol) (Note 10) is added dropwise over 15 min using a syringe while maintaining an internal temperature below −70 °C (Note 11). After being stirred for another 60 min at −78 °C, the mixture is allowed to warm to ambient temperature. After being stirred overnight (17 h), the reaction is quenched at ambient temperature with 1 N aq. hydrochloric acid (4 × 25 mL), and ethyl acetate (100 mL) is added (Note 12). The reaction mixture is transferred to a 1-L separatory funnel and the organic phase is collected. The aqueous phase is extracted with ethyl acetate (2 × 100 mL). The combined organic phases are washed with brine (100 mL). Drying the extracts with anhydrous sodium sulfate (35 g), filtration, and removal of the solvent under reduced pressure on a rotary evaporator (35 °C, 40 mmHg) afford the crude product as a light yellow solid (Note 13). The solid is transferred to a 500-mL, round-bottomed flask equipped with a reflux condenser. Ethyl acetate (60 mL) is added, and the solution is heated to reflux in a mineral oil bath (Note 14). Hexanes (150 mL) are added slowly through the condenser while maintaining the solution at reflux (Note 15). The resulting suspension is allowed to cool to room temperature and then placed in a freezer at −20 °C. After standing at −20 °C overnight, the crystals are collected by filtration and are rinsed with 40 mL of hexane/EtOAc, 4:1. The product is dried in a 100-mL round-bottomed flask fitted with a vacuum adapter and evacuating the flask (0.02 mmHg) for 24 h. Thus, 3.65 g (40%) of (2R,3R)-1,4-bis[(4S)-4-isopropyl-2-oxo-(1,3-oxazolidine-3-yl)]-2,3-dimethylbutane-1,4-dione (3) is obtained as white, cotton-like crystals (Notes 16 and 17).
C. (2R,3R)-2,3-Dimethylsuccinic acid (4). A 250-mL, single-necked, round-bottomed flask under an argon atmosphere equipped with a Teflon-coated magnetic stirring bar is charged with 3.68 g (10.0 mmol) of 3, THF (50 mL), and water (40 mL) (Note 18). After purging the reaction flask with argon, the resulting mixture is cooled to 0 °C (bath temperature) in an ice-water bath. To this solution is added via syringe 30% aq.hydrogen peroxide (11 mL, ~102 mmol) over 5 min (Note 19), followed by addition of lithium hydroxide monohydrate (1.68 g, 40 mmol) in one portion. The resulting solution is allowed to warm to ambient temperature. After being stirred for 18 h (Note 20), the reaction mixture is cooled to 0 °C (bath temperature) in an ice-water bath, and the reaction is quenched with 1.5 M aqueous Na2SO3 solution (40 mL). The resulting mixture is transferred to a 500-mL separatory funnel and is extracted with dichloromethane (3 × 80 mL) (Note 21). The aqueous layer is transferred to a 250-mL single-neck round-bottomed flask equipped with a Teflon-coated magnetic stirring bar and is cooled to 0 °C (bath temperature) in an ice-water bath. Concentrated aq. hydrochloric acid (12 M, 20 mL) is added over 5 min. The resulting solution is transferred to a 250-mL separatory funnel and is extracted with ethyl acetate (3 × 60 mL). The combined organic phases are washed with brine (100 mL). Drying the organic extracts with anhydrous sodium sulfate (15 g), filtration, and removal of the solvent under reduced pressure on a rotary evaporator (30 °C, 40 mm Hg) afford the crude product as a white solid. The product is dried in a 100-mL, round-bottomed flask fitted with a vacuum adapter and evacuating the flask (0.02 mmHg) for 24 h to afford 1.34 g, (92%) of 2R,3R)-2,3-dimethylsuccinic acid as a colorless solid (Note 22).
D. (2R,3R)-2,3-Dimethylbutane-1,4-diol (5). An oven-dried, 250-mL, three-necked, round-bottomed flask is equipped with a rubber septum, a 50-mL pressure-equalizing addition funnel fitted with a rubber septum, a reflux condenser fitted with an argon inlet adapter, and a Teflon-coated magnetic stirring bar. The flask is charged with 0.857 g (22.6 mmol) of lithium aluminum hydride and THF (56 mL) (Notes 1 and 23). (CAUTION: lithium aluminum hydride is very reactive and must be handled carefully to avoid contact with water). The resulting suspension is cooled to 0 °C (bath temperature) in an ice-water bath, and a solution of (2R,3R)-2,3- dimethylsuccinic acid (1.10 g, 7.53 mmol) in THF (24 mL) is added dropwise over 10 min from the dropping funnel (Note 24), which is rinsed with THF (4 mL). The resulting suspension is heated at reflux in a mineral oil bath for 12 h (Note 25). The reaction mixture is cooled to 0 °C (bath temperature) in an ice-water bath and is diluted with ethyl ether (80 mL). Water (0.9 mL) is added dropwise slowly (Note 26), followed after 5 min by 3 M aq. sodium hydroxide solution (0.9 mL) and, after 5 min, more water (2.6 mL). The resulting mixture is allowed to warm to ambient temperature and is stirred at this temperature for 2 h. The white precipitate is removed by filtration of the mixture through a ceramic Büchner funnel lined with filter paper (Whatman 90 mm thick qualitative circles) fitted onto a 100-mL filter flask. The white residue is transferred to a 50-mL, round-bottomed flask equipped with a Teflon-coated magnetic stirring bar. After breaking the precipitate into small pieces with a metal spatula, THF (20 mL) is added and then the suspension is heated at reflux in a mineral oil bath with vigorous stirring for 60 min (Note 27). The precipitate is removed by filtration through a medium-porosity, glass-fritted Büchner funnel into a 100-mL filter flask and is washed with ethyl ether (3 × 20 mL). The combined filtrates are dried with anhydrous sodium sulfate (6 g), filtered, and concentrated under reduced pressure on a rotary evaporator (25 °C, 40 mmHg) to afford the crude product as a light yellow oil (Note 28). The product is purified by Kugelrohr distillation (Note 29) to afford 0.80 g (90%) of (2R,3R)-2,3-dimethylbutane-1,4-diol as a viscous, clear oil (Notes 30, 31, and 32).
2. Notes
1.
The checkers obtained tetrahydrofuran by filtration through an alumina column under a N
2 atmosphere on a GlassContour system. The submitters distilled tetrahydrofuran from sodium benzophenone ketyl under an atmosphere of argon.
2.
The checkers purchased
(4S)-isopropyl-2-oxazolidinone from Acros, Inc. The submitters purchased
(4S)-isopropyl-2-oxazolidinone from Aldrich Chemical Company, Inc. or prepared it from L-valine according to the following procedure (the chemicals were purchased from Aldrich Chemical Company, Inc. and were used as received):
A)
Reduction of L-valine to (S)-valinol with LiAlH4 according to the procedure reported in reference 2:
L-valine (40.0 g, 340 mmol) was added slowly, in small portions, to a two-neck 2-L flask equipped with a Teflon-coated magnetic stirring bar, a reflux condenser, and an an argon inlet adapter on top of the condenser, containing a mixture of
lithium aluminum hydride (25.9 g, 680 mol) in
800 mL of dry THF (Note 1) cooled to 0 °C with an ice-water bath. After all of the L-valine had been added (about 1 h), the reaction mixture was heated at reflux for 10 hours. The solution was cooled to 0 °C, and excess LiAlH
4 was quenched with
100 mL of aq. sodium hydroxide (2.0 M). The precipitate was filtered off and extracted with boiling
THF (100 mL) for an hour and filtered off again. The combined organic filtrates were dried with sodium sulfate, filtered, and the solvent was removed under reduced pressure on a rotary evaporator (20 mmHg) to give
28.5 g of (S)-valinol (276 mmol, 81% yield) as a colorless oil, which was used in the next step without further purification.
B)
Preparation of (4S)-isopropyl-2-oxazolidinone from (S)-valinol and diethyl carbonate according to the procedure in reference 3: A 100-mL flask equipped with a 10-cm Vigreux column and a Teflon-coated magnetic stirring bar was charged with
(S)-valinol (28.5 g, 276 mmol),
diethyl carbonate (36.8 mL, 304 mmol), and
anhydrous potassium carbonate (3.87 g, 28 mmol). The mixture was heated at 135 °C (temperature of the oil bath) until no more ethanol distilled (32 mL EtOH was distilled off after 5 h of heating). The resultant mixture was cooled to room temperature and dissolved in
diethyl ether (800 mL), and the solution was filtered through a 2-cm pad of Celite to remove the potassium carbonate. The filtrate was concentrated under reduced pressure on a rotary evaporator (20 mmHg) to 250 mL and then was placed into a 0 °C refrigerator. After 12 h, the crystals were collected by filtration. The filtrate was concentrated to 60 mL and placed in a 0 °C refrigerator overnight followed by filtration to provide additional product. The combined crystals were dried under vacuum to afford
(4S)-isopropyl-2-oxazolidinone 29.9 g (232 mmol, 84% yield) as white needles.
3.
n-Butyllithium in hexane was purchased from Acros Chemical Company, Inc. and was titrated according to the method of Gilman.
44.
Flash chromatography was performed by using an 8-cm diameter column packed with 250 g of silica gel (MP Silitech 32-63 D 60 Å). The product was eluted with 2 L of hexane/EtOAc, 17:3, followed by 1 L of hexane/EtOAc, 4:1, and 0.5 L of hexane/EtOAc, 3:1. After collection of
500 mL of eluent, 50 mL-fractions were collected. The submitters report that the product can be purified by distillation at 102–106 °C/0.75 mmHg. The checkers found that distillation did not successfully separate remaining starting material from the product.
5.
Thin layer chromatography (TLC) analysis is used to identify product fractions. TLCs are performed by using Merck silica gel 60 F
254 analytical plates; detection with UV or by dipping in a KMnO
4 solution which was prepared from
KMnO4 (3 g),
K2CO3 (20 g),
NaOH 5% (5 mL) in
H2O (300 mL) followed by heating. Product R
f = 0.38 and starting material R
f = 0.06 in hexane/EtOAc, 7:3.
6.
To ensure complete removal of ethyl acetate and hexanes, anhydrous toluene (2 × 20 mL; obtained by filtration through a drying column on a GlassContour system) is added to the purified product and concentrated by rotary evaporation followed by drying under high vacuum (0.02 mm Hg) for 12 h.
7.
The product,
(4S)-isopropyl-3-propionyl-2-oxazolidinone, is commercially available from Aldrich Chemical Company. The procedure provided here is based on the procedure in reference 5a. The product displayed the following physicochemical properties: [α]
D20 +96.3 (CH
2Cl
2, c 1.0), lit.
5b [α]
D20 +96.8 (CHCl
3, c 8.7);
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.87 (d,
J = 7.0 Hz, 3 H), 0.91 (d,
J = 7.0 Hz, 3 H), 1.17 (t,
J = 7.3 Hz, 3 H), 2.31-2.44 (m, 1 H), 2.84-3.04 (m, 2 H), 4.20 (dd,
J = 3.0 Hz, 9.0 Hz, 1 H), 4.24 (at,
J = 8.4 Hz, 1 H), 4.43 (dt,
J = 8.4 Hz, 3.3 Hz, 1 H).
13C NMR
pdf (100 MHz, CDCl
3) δ: 8.5, 14.8, 18.1, 28.5, 29.3, 58.5, 63.5, 154.3, 174.2. HRMS (FAB)
m/z: calcd. for C
9H
16NO
3 [M+H] 186.1130, found 186.1128.
8.
The checkers used an egg-shaped Fisherbrand 19 mm × 41 mm stirring bar.
9.
Diisopropylamine was purchased from Aldrich Chemical Company, Inc. and was distilled under nitrogen (atmospheric pressure) from calcium hydride prior to use.
10.
Titanium (IV) chloride was purchased from Aldrich Chemical Company, Inc. and was used as received.
11.
The reaction mixture becomes viscous as titanium chloride is added at low temperature, therefore it is important to ensure that efficient stirring is maintained during the addition of the titanium reagent. The reaction mixture turns dark purple and yellow smoke forms upon addition of the titanium chloride. The smoke thickens during the progress of the titanium chloride addition, and disappears 5–10 min after the addition is complete.
12.
Aqueous hydrochloric acid (37%, ~12 N) certified A.C.S. PLUS was purchased from Fisher Chemical Fisher Scientific and diluted to the concentration of 1 N with water. The 1 N HCl is added in four portions with 1 min intervals while maintaining the reaction temperature below 40 °C.
13.
The submitters noted that
1H NMR analysis of the crude product mixture indicated 65–70% yield of the homocoupling product, the remainder of the material being mostly the starting propionyl oxazolidinone. Complete conversion was never achieved under a variety of reaction conditions.
14.
The solid fully dissolves upon bringing the solution to reflux.
15.
The product precipitates during the addition of hexanes.
16.
The product has the following physicochemical properties: mp 249–250 °C (lit.
6 mp 252–253 °C); [α]
D20 +40.3 (CH
2Cl
2, c 1.0), lit.
6 [α]
D20 +40.5 (CHCl
3, c 1.0);
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.84-0.92 (m, 12 H), 1.22 (d,
J = 6.0 Hz, 6 H), 2.19-2.32 (m, 2 H), 4.00-4.12 (m, 2 H), 4.20 (dd,
J = 3.0, 9.0 Hz, 2 H), 4.26 (at,
J = 8.6 Hz, 2 H), 4.36 (dt,
J = 8.4, 3.0 Hz, 2 H).
13C NMR
pdf (100 MHz, CDCl
3) δ: 14.5, 15.2, 18.1, 28.1, 41.4, 58.8, 63.1, 153.6, 177.2; IR (film) cm
−1: 2970, 1768, 1691, 1384, 1232, 1194, 1099; HRMS (FAB)
m/z: calcd for C
18H
28N
2O
6Na [M+Na] 391.1845, found 391.1854. Anal. Calcd. for C
18H
28N
2O
6: C, 58.68; H, 7.66; N, 7.60. Found: C, 58.73; H, 7.84; N, 7.60.
17.
On half scale
(25 mmol), the checkers obtained 2.57 g (56% yield) of (2R,3R)-1,4-bis[(4S)-4-isopropyl-2-oxo-(1,3-oxazolidine-3-yl)]-2,3-dimethylbutane-1,4-dione.
18.
The (4
S)-isopropyl-3-propionyl-2-oxazolidone does not fully dissolve in THF and a thick slurry forms upon addition of the water.
19.
30%
Hydrogen peroxide was purchased from Aldrich Chemical Company, Inc. and was used as received.
20.
Reaction progress was monitored by the disappearance of starting material by thin layer chromatography (TLC). TLC analysis was performed using Merck silica gel 60 F
254 analytical plates; detection with UV or by dipping in a KMnO
4 solution, which was prepared from
KMnO4 (3 g),
K2CO3 (20 g),
NaOH 5% (5 mL) in
H2O (300 mL) followed by heating. Starting material R
f = 0.6 with CH
2Cl
2/MeOH, 20:1 as the eluent (the product remained at the baseline under these elution conditions).
21.
The combined organic phases are washed with
brine (100 mL). Drying the organic extracts over anhydrous sodium sulfate, filtration, and removal of the solvent under reduced pressure on a rotary evaporator (40 mmHg) affords the recovered
(4S)-(-)-isopropyloxazolidin-2-one as a crystalline solid (2.30 g, 89%).
22.
The product has the following physicochemical properties: mp 130–131 °C (lit.
7 mp 134–135 °C); [α]
D20 +7.4 (H
2O, c 1.0), (lit.
7 [α]
D20 +8.4 (H
2O, c 4.0);
1H NMR
pdf (400 MHz, CD
3OD) δ: 1.16 (d,
J = 6.8 Hz, 6 H), 2.68-2.80 (m, 2 H), 4.92 (br s, 2 H);
13C NMR
pdf (100 MHz, CD
3OD) δ: 14.0, 42.8, 179.1; IR (film) cm
−1: 2982, 2909, 1701, 1461, 1419, 1283, 1213, 932; HRMS (FAB)
m/z: calcd. for C
6H
11O
4 [M+H] 147.0657, found 147.0658. Anal. Calcd. for C
6H
10O
4: C, 49.31; H, 6.90. Found: C, 49.46; H, 7.01.
23.
Lithium aluminum hydride was purchased from Aldrich Chemical Company, Inc. and was used as received.
24.
The checkers observed vigorous bubbling upon addition of (2
R,3
R)-2,3- dimethylsuccinic acid.
25.
Reaction progress was monitored by thin layer chromatography (TLC). TLC analysis was performed using Merck silica gel 60 F
254 analytical plates; detection by dipping in a solution of bromocresol green [prepared by dissolving
bromocresol green (0.04 g) in
EtOH (100 mL) and slowly dripping in a 0.1 M solution of NaOH until the solution turns pale blue]. Starting material R
f = 0.1 (yellow) and product R
f = 0.2 (white) with CH
2Cl
2/MeOH, 20:1 as the eluent.
26.
Water reacts violently with lithium aluminum hydride (LAH) necessitating slow addition of the water and cooling with a 0 °C ice-water bath.
27.
This procedure extracts any product trapped in the white precipitate. The majority of the product (~ 80%) is in the filtrate.
28.
The product has moderate volatility due to its low molecular weight. Care must therefore be taken to minimize loss during concentration. No loss was observed during concentration on a rotary evaporator (40 mmHg), but the product should not be placed under high vacuum for extended time periods.
29.
The distillation was performed at 110 °C, 10 mmHg. A 10-mL collection flask was cooled with an ice-water cooling bath.
30.
The product has the following physicochemical properties: bp 105–110 °C, 10 mmHg. [α]
D20 +10.4 (CH
2Cl
2, c 1.0), (lit.
8 [α]
D20 +8.0 (CH
2Cl
2, c 1.0)).
1H NMR
pdf (500 MHz, CDCl
3) δ: 0.91 (d,
J = 6.8 Hz, 6 H), 1.66-1.75 (m, 2 H), 2.40 (br s, 2 H), 3.52 (dd,
J = 10.8, 5.8 Hz, 2 H), 3.64 (dd,
J = 10.8, 3.8 Hz, 2 H).
13C NMR
pdf (125 MHz, CDCl
3) δ: 13.8, 37.8, 65.9; IR (film) cm
−1: 3274, 2959, 2920, 2871, 1452, 1384, 1040; HRMS (FAB)
m/z: calcd for C
6H
15O
2 [M+H] 119.1072, found 119.1072. Anal. Calcd. for C
6H
14O
2, C, 60.98; H, 11.94. Found: C, 60.76; H, 11.79.
31.
The diastereomeric purity was determined by the checkers to be >98% by 125 MHz
13C NMR (S/N = 46:1) and comparison to literature values for the meso diol.
9 13C NMR (125 MHz, CDCl
3) δ: 13.6, 38.9, 65.5 for the meso diol.
32.
The checkers established enantiomeric purity by HPLC analysis of the (
R)-MTPA and (
S)-MTPA diesters, [prepared according to the procedure reported in reference 10 using
(2R,3R)-2,3-dimethylbutane-1,4-diol> (3.0 mg, 0.025 mmol),
MTPA-Cl (0.038 mL, 0.20 mmol),
Et3N (0.035 mL, 0.25 mmol), and
DMAP (25 mg, 0.20 mmol), in
CH2Cl2 (0.12 mL, 0.2M) at rt for 12 h]. The enantiomeric ratio was determined to be 99.2:0.8 by HPLC analysis using an Agilent 1100 series LC equipped with a silica normal phase column (Microsorb Si 100 Å packing) with a multiwavelength detector (hexanes/
tert-butyl methyl ether, 97:3, 1 mL/min, λ = 210 nm,
tR (
R, R, R, R) = 37.7 min,
tR (
S, R, R, S) = 38.9 min). The submitters established enantiomeric purity by comparison of the 500 MHz
1H NMR spectrum of the (
R)-MTPA diester derivative with the
1H NMR spectrum of a sample prepared from racemic 2,3-dimethylsuccinic acid (Aldrich). A comparison of the vicinal methyl groups was the most informative and indicated that none of the minor enantiomer could be detected.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
(2
R,3
R)-2,3-Dimethyl-1,4-butanediol is a valuable
C2-symmetric building block that has potential utility for the preparation of novel asymmetric catalysts and has been employed in studies related to natural product synthesis.[KB1] (2
R,3
R)-2,3-Dimethylsuccinic acid, which serves as an intermediate in its preparation, is also of value. Several enantioselective syntheses of the diol and the diacid have been documented in the literature. One of the earliest is a report by McCasland and Proskow that is based on resolution of racemic dimethylsuccinic acid by fractional crystallization of its dibasic salt with brucine.
7 The diacid was subsequently reduced to the diol via its dimethyl diester (Scheme 1). Recently, this procedure was adopted by Widenhoefer and co-workers to access (2
R,3
R)-2,3-dimethyl-1,4-butanediol via (2
R,3
R)-2,3-dimethylsuccinic acid in 62% ee.
11 The somewhat tedious fractional recrystallization and poor availability of racemic 2,3-dimethylsuccinic acid
12 make this method less appealing.
13Scheme 1
Chan and co-workers developed a method capitalizing on diastereoselective alkylation of chiral
N-acylnorbornene sultams with alpha-haloesters that is suitable for the synthesis of (2
R,3
R)-2,3-dimethyl-1,4-butanediol (Scheme 2).
14 Although this approach leads to highly enantiopure (2
R,3
R)-2,3-dimethyl-1,4-butanediol, multistep preparation of the requisite chiral auxiliary, which requires chromatographic separations, significantly detracts from the practicality of this method.
Scheme 2
Another chiral auxiliary method suitable for the preparation of the diol has been reported by Feringa and co-workers (Scheme 3).
15 Sequential treatment of 5-(
l-Menthyloxy)-2(5
H)-furanone with tri(methylthio)methyl-lithium and iodomethane at −90 °C affords butenolide
1, which is desulfurized with Raney Ni and then reduced with lithium aluminum hydride to provide enantiopure (2
R,3
R)-2,3-dimethyl-1,4-butanediol.
Scheme 3
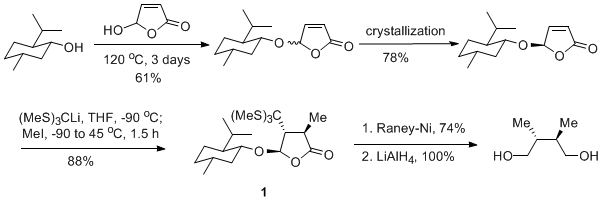
Oxidative homocoupling of lithiated chiral
N-acyloxazolidinones described by Kise and co-workers provided the basis for the procedure described here. It appears to be the most direct method for the synthesis of both enantioenriched (2
R,3
R)-2,3-dimethyl-1,4-butanediol and (2
R,3
R)-dimethylsuccinic acid (Scheme 4). Some of the advantages of this approach are the wide availability of the Evans chiral auxiliary, which serves as the source of chirality in this case, and the experimental simplicity of the reactions. Titanium tetrachloride, CuCl
2, PhI(OAc)
2, and iodine have been employed as oxidants in the homocoupling reaction. After careful screening, we found that TiCl
4 gives the most consistent results and yields compared to those obtained with the other reagents. Although Kise and co-workers reported yields in the range of 65–68%, we reproducibly obtained the homocoupling product in 40–45% yield after recrystallization.
16 The procedure employing TiCl
4 has also proven to be the most convenient experimentally.
Scheme 4
Subsequent removal of the chiral auxiliary with lithium hydroperoxide, according to the protocol developed by Evans and co-workers,
17 and direct reduction of the diacid with lithium aluminum hydride delivered (2
R,3
R)-2,3-dimethyl-1,4-butanediol of high enantiomeric purity (>99:1 er, 30% overall yield) as determined by 500 MHz
1H NMR spectroscopy of the derived (
R)-Mosher diester and analytical HPLC. The overall preparation requires only four steps from commercially available starting materials and a single chromatographic purification.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(4S)-Isopropyl-2-oxazolidinone:
(4S)-4-(1-Methylethyl)-2-oxazolidinone; (17016-83-0)
Propionyl chloride:
Propanoyl chloride; (79-03-8)
n-Butyllithium; (109-72-8)
(4S)-Isopropyl-3-propionyl-2-oxazolidinone:
(4S)-4-(1-Methylethyl)-3-(1-oxopropyl)-2-oxazolidinone; (77877-19-1)
(2R,3R)-1,4-Bis[(4S)-4-isopropyl-2-oxo-(1,3-oxazolidine-3-yl)]-2,3-dimethylbutane-1,4-dione:
(4S,4'S)-3,3'-[(2R,3R)-2,3-Dimethyl-1,4-dioxo-1,4-butanediyl]bis[4-(1-methylethyl)-2-oxazolidinone; (259540-48-2)
Diisopropylamine:
2-Propanamine, N-(1-methylethyl)-; (108-18-9)
Titanium (IV) chloride (7550-45-0)
(2R,3R)-2,3-Dimethylsuccinic acid; (5866-39-7)
Hydrogen peroxide; (7722-84-1)
(2R,3R)-2,3-Dimethylbutane-1,4-diol:
(2R,3R) 2,3-Dimethyl-1,4-butanediol; (127253-15-0)
Lithium aluminum hydride; (16853-85-3)
 |
Armen Zakarian was born in Moscow in 1973 and completed his undergraduate studies at Moscow State University in 1994. His Diploma research was completed at the Zelinsky Institute of Organic Chemistry with Dr. Vladimir Borodkin. He received his Ph.D. under the direction of Professor Robert A. Holton at Florida State University in 2001, and then spent two years (2002–2004) in the laboratories of Professor Larry E. Overman (University of California, Irvine) as a postdoctoral research associate. In August 2004, he joined the faculty at the Department of Chemistry and Biochemistry, Florida State University, and will be moving to the University of California, Santa Barbara in 2008. His research interests include the total synthesis of natural products, bioorganic chemistry, and the development of synthetic methodology.
|
 |
Chongdao Lu was born in 1979 and carried out his undergraduate studies at Yunnan University (1996–2000). He received his doctorate degree in 2005 from Chengdu Institute of Organic Chemistry, working under the direction of Professors Wen-Hao Hu and Ai-Qiao Mi on novel methodology involving ylide chemistry. In 2005, he joined the group of Armen Zakarian, where he is a postdoctoral research associate. His current interests are the total synthesis of natural products and the development of new methodology for organic synthesis. He is a recipient of the MDS Postdoctoral Fellowship (2006-present).
|
 |
Katrien Brak was born in 1983 in Leuven, Belgium. She earned a B.S. degree in chemistry from the Massachusetts Institute of Technology in 2005. That year, she began her doctoral studies at the University of California, Berkeley in the laboratories of Prof. Jonathan A. Ellman. Her graduate work has focused on the development of nonpeptidic protease inhibitors as well as the development of methods for the asymmetric synthesis of amines.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved