Org. Synth. 2008, 85, 248
DOI: 10.15227/orgsyn.085.0248
(R)-3-METHYL-3-PHENYL-1-PENTENE VIA CATALYTIC ASYMMETRIC HYDROVINYLATION
Submitted by Craig R. Smith, Aibin Zhang, Daniel J. Mans, and T. V. RajanBabu
1,2.
Checked by Scott E. Denmark and Min Xie.
1. Procedure
Caution!
Grignard reagents similar to 3,5-bis(trifluoromethyl)phenylmagnesium bromide have been known to decompose violently,3 hence it is suggested that the reaction be conducted behind a blast shield in an efficient fume hood.
A.
Sodium Tetrakis[(3,5-trifluoromethyl)phenyl]borate (NaBArF24) (2).3–6 NaBArF
24 can be conveniently prepared following the
Inorganic Syntheses procedure.
4b Although this procedure is reproducible, the additional details and the minor modifications described below are recommended.
Following the procedure described in
Inorganic Syntheses,4b a Grignard reaction employing Mg turnings (1.17 g, 48.1 mmol, 6.5 equiv),
sodium tetrafluoroborate (813 mg, 7.40 mmol),
1,2-dibromoethane (0.57 mL, 1.25 g, 6.7 mmol, 0.9 equiv) and
3,5-bis(trifluoromethyl)bromobenzene (7.15 mL, 12.16 g, 41.5 mmol, 5.6 equiv) (Note 1) is carried out in anhydrous ether
(Note 2) under an argon atmosphere, The reaction mixture is stirred at room temperature for 12 h to ensure the completion of reaction. After the workup and the subsequent processing of the reaction mixture as described ref 4b, the solvent is removed from the ether extracts using a rotary evaporator (20 °C, ca. 20 mmHg). The resulting heavy, brown oil is dried by azeotropic distillation using a Dean-Stark trap and 200 mL of dry benzene
(Note 3) under argon for two hours. Most
(>190 mL) of the benzene is then removed by a continuously draining the solvent through the stopcock on the Dean-Stark trap
(Note 4). After being cooled to room temperature, the remaining oily, brown mixture is triturated with
dry benzene (50 mL) and the benzene layer is then decanted. The trituration is repeated twice more or until the resulting benzene layer is colorless
(Note 5). The residual solvent is removed from the mixture with a rotary evaporator (ca. 40 °C, 20 mmHg). The remaining tan solid is further dried under high vacuum (0.1 mmHg) at room temperature for 8 h to afford a light tan powder (6.15 g, 94%)
(Note 6). To further purify the product, the tan powder is transferred to a ceramic Büchner funnel (60 mm × 100 mm (d × h)) fitted with a Whatman 1 filter paper (55 mm) where it is washed with three, 30-mL portions of a cold (−40 °C) mixture of dichloromethane/hexane, 2:1
(Note 7) in a Büchner funnel to remove colored impurities. The solid is then dried under vacuum (90 °C, 0.1 mmHg) for 16 h and then is cooled to ambient temperature to afford 4.96 g (76%) of the sodium salt as a free-flowing, fine, white powder (Notes
8 and
9). Typically, the salt is stored at ambient temperature in a glove-box.
B.
Di(μ-bromo)bis(η-allyl)nickel(II) (4).7,8 A 250-mL, three-necked, round-bottomed flask equipped with a rubber septum, a Teflon-taped flow-controlled argon inlet, a thermometer, and a magnetic stirring bar is flame-dried, purged with argon and transferred into a glove-box. The flask is then charged with
bis[1,2:5,6-η-(1,5-cyclooctadiene)]nickel (1.38 g, 5.00 mmol) (Note 10) and is removed from the glove-box. Under argon from a Schlenk line, the flask is charged with
anhydrous diethyl ether (80 mL) (Note 11) and is cooled to −70 °C in a dry ice/2-propanol bath, at which time allyl
bromide (0.440 mL, 0.615 g, 5.08 mmol, 1.0 equiv) (Note 12) is added
via syringe with stirring. The cold bath is removed and replaced by a ice-water bath. The reaction mixture is allowed to warm to 0 °C gradually (ca. 15 min), then to ambient temperature over another 15 min by removing the cold bath
(Note 13). The reaction mixture is stirred at ambient temperature for another 30 min and then is transferred into the glove-box. The volatile contents of the flask are removed under high vacuum (ca. 0.1 mmHg) by connecting the flask to an evacuation port inside the glove-box
(Note 14). The flask is rinsed with
anhydrous ether (3 × 10 mL) and the resulting suspension is passed through a plug of dry Celite (2.5 cm × 7.5 cm (d × h)) in a fritted glass filter funnel (2.5 cm × 18 cm, (d × h) coarse)
(Note 15). The Celite pad is washed with an additional 40 mL of ether. The ether washings are transferred into a 250-mL pear-shaped flask equipped with a magnetic stir bar. The ether is removed under vacuum (ca. 0.1 mmHg) with vigorous stirring to afford 0.803 g (89%) of the title compound as a dark-red powder, which was used without further purification (Note
16,
17).
C. 2-Phenyl-1-butene (6). A 2-L, two-necked, round-bottomed flask equipped with a condenser (fitted with a nitrogen inlet), a rubber septum, and a magnetic stir bar is evacuated, flame-dried, and purged with nitrogen. The flask is then charged with methyltriphenylphosphonium bromide (43.4 g, 121 mmol, 1.2 equiv) (Note 18) through one neck of the flask under a strong flow of nitrogen and then quickly sealed with the rubber septum. Anhydrous THF (500 mL) (Note 19) is added into the flask through the rubber septum. The contents of the flask are then treated with n-BuLi (76.0 mL, 1.6 M solution in hexanes, 121 mmol, 1.2 equiv) (Note 20) at ambient temperature and the mixture is stirred for 2 h. Propiophenone (13.4 mL, 101 mmol) (Note 21) is then added dropwise via syringe over a period of 15 min. The reaction mixture is heated to reflux with stirring for 8 h. The mixture is allowed to cool to ambient temperature and 400 mL of hexane/diethyl ether, 1:1 (Note 22) is added. The resulting suspension is filtered through a pad of Celite (ca. 15 g, 5 cm × 5 cm) in a fritted glass filter funnel [5.0 cm × 12 cm, (d × h) coarse] which is washed with 100 mL of hexane/diethyl ether, 1:1 (Note 23). The filtrate is concentrated by rotary evaporation (15 °C, ca. 20 mmHg). The resulting pale-yellow slurry is first taken up in dichloromethane (30 mL) and then is triturated with hexane (200 mL) and the supernatant liquid is decanted. The remaining slurry is treated in the same manner twice more (Note 24). The combined extracts are filtered through a silica plug (ca. 20 g, 3 cm long, 5 cm in diameter), which is washed with 50 mL of hexane. The combined eluates are concentrated by rotary evaporation (15 °C, ca. 20 mmHg). The light-yellow liquid residue is purified by column chromatography (Note 25) to afford 11.9 g (88%) of the 6 as a clear colorless liquid (Note 26).
D.
(R)-3-Methyl-3-phenylpentene (7).
2 The pre-catalyst is prepared as follows
in a glove-box: A 100-mL, pear-shaped Schlenk flask with one side-arm fitted with a rubber septum and equipped with a magnetic stirring bar is evacuated, flame-dried, and purged with argon. The flask is charged with
anhydrous dichloromethane (50 mL) (Note 27) and is transferred into a glove-box
(Note 28). To the flask is quickly added
[di(μ-bromo)bis(η-allyl)nickel(II) (180 mg, 0.50 mmol, 0.01 equiv),
(R)-2,2'-binaphthoyl-(S,S)-di(1-phenylethyl)aminoylphosphine (539 mg, 1.00 mmol, 0.02 equiv) (Note 29) and
NaBArF24 (886 mg, 1.00 mmol, 0.02 equiv) in the order mentioned. The resulting suspension is stirred at ambient temperature for 2 h to afford a dark-brown solution containing a small amount of fine particles (NaBr).
A 1-L, three-necked, round-bottomed flask equipped with a rubber septum, a Teflon-taped flow-controlled argon inlet, a thermometer, and a magnetic stir bar is flame-dried and purged with argon. The flask is then charged with 150 mL of anhydrous dichloromethane (Note 27). The catalyst solution prepared above, now removed from the drybox, is introduced to the vessel via cannula. The flask containing the catalyst solution is further rinsed with 10 mL of dichloromethane, and this solution is also transferred to the reaction mixture. Upon completion of pre-catalyst transfer, the system closed at the flow-controlled stopcock and then is cooled to −70 °C in a dry ice/acetone bath, creating a small vacuum. A strong flow of dry ethylene (Note 30) is introduced through a needle through the serum stopper to relieve the vacuum and then is adjusted to maintain a pressure of 1 atm by releasing excess gas through an oil bubbler. The introduction of the ethylene causes the internal temperature to rise. Within ca. 5 min, the internal temperature increases by 5 °C and the ethylene line is removed (Note 31). The solution is cooled back to −70 °C with vigorous stirring. A solution of 2-phenyl-1-butene (6) (6.60 g, 50.0 mmol) in 30 mL of dry dichloromethane (Note 27) is introduced as a weak stream into the solution of pre-catalyst over a two-minute period via syringe followed by a 10 mL rinse with dichloromethane. Ethylene is introduced again through a needle, first as a strong flow and then regulated to maintain a pressure of 1 atm. Under an ethylene atmosphere, the internal temperature of the reaction mixture is then maintained between −65 °C and −70 °C for a period of 4 h. At the end of this period the ethylene line is removed and the reaction mixture is slowly poured into an Erlenmeyer flask containing 500 mL of pentane (Note 32) is and combined with a 50-mL pentane rinse of the reaction vessel. After being warmed to ambient temperature, the resulting, cloudy solution is filtered through a plug of silica gel (Merck, grade 9385, mesh 230-400, 60 Å, 4 cm × 5 cm, (d × h)), which is eluted with 100 mL of pentane (Note 33). The combined eluates are concentrated by rotary evaporation (20 °C, 20 mmHg) to afford 7.99 g (99.7%) of (R)-3-methyl-3-phenylpentene (er 98.8:1.2) as a clear, liquid (Notes 34 and 35).
2. Notes
1.
Reagent-grade magnesium turnings (99.98%) were purchased from Reade Manufacturing Company and activated prior to use with 1,2-dibromoethane following the method described by Reger.
4b Sodium tetrafluoroborate (98%, Sigma-Aldrich Company) was dried at 100 °C under vacuum (0.1 mmHg) for 12 h and then was cooled to ambient temperature before being transferred into a glove-box. It was kept at ambient temperature in a glove-box for prolonged storage.
1,2-Dibromoethane (Sigma Chemical Company) was dried over 4 Å molecular sieves overnight before usage.
3,5-Bis(trifluoromethyl)bromobenzene (98%) was purchased from Matrix Scientifics and was used as received.
2.
Diethyl ether (Fisher, BHT stabilized, HPLC grade) was dried by percolation through two columns packed with neutral alumina under a positive pressure of argon.
3.
Benzene (Sigma-Aldrich Company) was distilled from sodium ribbon under a nitrogen atmosphere.
4.
The benzene was not completely removed at this stage while the flask was still warm.
5.
Benzene soluble impurities removed at this stage and a beige solid is formed.
6.
On a similar scale (
703 mg, 6.37 mmol of sodium tetrafluoroborate as the limiting reagent), 5.15 g (91%) of the salt was obtained at this stage.
7.
Reagent grade
dichloromethane (Sigma-Aldrich Company) and reagent grade
hexane (Fisher Scientific Company) were mixed and cooled to −40 °C using a dry ice-acetonitrile cold bath.
8.
On a similar scale (
703 mg, 6.37 mmol of sodium tetrafluoroborate was employed as the limiting reagent), 4.07 g (72%) of the salt was obtained.
9.
The product displayed the following physicochemical properties:
1H NMR
pdf (500 MHz, acetone-
d6) δ: 7.79 (br s, 8 H), 7.67 (br s, 4 H).
13C NMR
pdf (125 MHz, acetone-
d6) δ: 162.3 (q,
J = 49.8 Hz), 135.2 (s), 129.6 (qq,
J = 31.3, 2.9 Hz), 125.0 (q,
J = 272.4 Hz), 118.1 (s);
19F NMR (470 MHz, acetone-
d6) δ: -63.6; IR (KBr) cm
−1: 1781, 1712, 1628, 1356, 1282, 1130, 945, 932, 887, 838, 743, 710, 682, 670. Although the melting point of the title compound has been reported,
4b,6 the checkers found that the product thus obtained did not melt, but only gradually darkened when heated to above 300 °C and rapidly decomposed at 400 °C. Obtaining correct elemental analysis of highly fluorinated compounds, which do not burn completely, is difficult. Although the purity of the title product could not be secured by elemental analysis, it was sufficiently pure to be employed in the generation of the pre-catalyst (allyl)Ni(phosphine) BARF.
10.
The
bis[1,2:5,6-η-(1,5-cyclooctadiene)]nickel was purchased from Strem Chemicals, Inc. and was used as received.
11.
Reagent-grade diethyl ether (Fisher Scientific Company) was freshly distilled from sodium benzophenone ketyl under an atmosphere of nitrogen prior to use.
12.
Allyl bromide (99%) was purchased from Sigma-Aldrich Company and was freshly distilled before use.
13.
The yellow Ni(COD)
2 crystals dissolve as the temperature approaches −10 °C, accompanied by the solution becoming pale orange. At 5 °C the solution turns deep red and becomes homogenous.
14.
To ensure the removal of all volatile components, the evacuation was continued for a period more than 4 h, during which time the vacuum was released temporarily every hour and the reaction vessel was weighed. The evacuation was resumed until the loss of mass was less than 10 mg between two consecutive measurements.
15.
The
Celite (Fisher Scientific Company) was first stirred with concentrated HCl overnight, and then was washed with deionized water until neutral and finally with reagent grade
MeOH (Sigma-Aldrich Company). The wet Celite was dried at 200 °C in an oven for 8 h and then was cooled to ambient temperature under vacuum (~0.1 mmHg). The dry Celite thus obtained was transferred into a glove-box before use.
16.
To ensure the complete removal of ether, the evacuation was continued for ca. 4 h, during which the weight of flask containing the product was checked every hour until the loss of mass was less than 5 mg.
17.
The allylnickel bromide dimer [di(μ-bromo)bis(η-allyl)nickel(II)] decomposes upon standing in air for several minutes. In an inert atmosphere, it is stable at 20 °C for several hours, however, it should be prepared in a drybox and stored at −20 °C in the freezer compartment if prolonged storage is required. No criterion of purity for this compound was established. This is a known compound
7,8 and several phosphine complexes of the allylnickel halides have been prepared by treatment with phosphines, and these complexes fully analyzed by
31P NMR,
1H NMR and X-ray crystallography.
14b However, experience validates the use of the crude material for further reactions as prescribed in this procedure.
18.
Methyltriphenylphosphonium bromide (99%) was purchased from Fisher Scientific Company and was used without further purification.
19.
THF (Fisher, BHT stabilized, HPLC grade) was dried by percolation through two columns packed with neutral alumina under a positive pressure of argon.
20.
n-BuLi was obtained as a solution in hexanes from Acros Organics and was titrated with
N-benzylbenzamide, 99%, (Acros Organics) before use.
21.
Propiophenone (99%) was purchased from Sigma Chemical Company and was used as received.
22.
Reagent grade
diethyl ether (Fisher Scientific Company) and ACS grade
hexane (Fisher Scientific Company) were used as received.
23.
The precipitated lithium bromide was thus removed.
24.
The title product isolated at this stage is only slightly contaminated by triphenylphosphine oxide, as confirmed by
1H NMR analysis of crude product.
25.
Column chromatography was performed with silica gel (Merck, grade 9385, mesh 230-400, 60 Å), column size, 9 cm × 6 cm (h × d). The product (
6) was isolated from 8 fractions (50 mL). UV and KMnO
4 detection were employed to visualize the product by TLC (silica gel); R
f value in pentane is 0.61. The product was sufficiently pure for use in the next stage. An analytically pure sample was obtained by a fractional distillation of a portion of the product at 45 mmHg and collecting the fraction boiling at 92–93 °C.
26.
The distilled product displayed the following physicochemical properties:
1H NMR
pdf (500 MHz, CDCl
3) δ: 7.43-7.41 (m, 2 H), 7.34-7.31 (m, 2 H), 7.29-7.24 (m, 1 H), 5.28 (s, 1 H), 5.12 (s, 1 H), 2.53 (q,
J = 7.3 Hz, 2 H), 1.17 (t,
J = 7.3 Hz, 3 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 150.0, 141.5, 128.2, 127.2, 126.0, 110.9, 28.0, 12.9; IR (neat) cm
−1: 3081, 3056, 3030, 2967, 2934, 2876, 1945, 1877, 1799, 1628, 1600, 1573, 1495, 1463, 1443, 894, 776, 702. Anal. Calcd. for C
10H
12: C, 90.85; H, 9.15. Found: C, 90.77; H, 9.15.
27.
Dichloromethane (Fisher, HPLC grade) was dried by percolation through two columns packed with neutral alumina under a positive pressure of argon.
28.
Because halogenated solvents can poison the catalysts in the glove box, it is recommended that the purifier of the glove box is temporarily turned off during the preparation of the dichloromethane solution of the pre-catalyst. A minimum exposure of dichloromethane to the atmosphere in the glove box is also recommended. The residual dichloromethane vapor can be removed by purging the glove box with argon before turning the purifier back on.
29.
See previous procedure in this volume.
30.
Ethylene (99%) was purchased from Matheson Tri-gas. The gas flow was controlled by a regulator. The ethylene line was split into two lines by a three way stopcock, one connected to an oil bubbler, and the other to a 10 cm × 1.3 cm column of Drierite® in front of a needle outlet.
31.
The checkers observed that if the exotherm was allowed to continue, the internal temperature of the reaction mixture could not be lowered to −70 °C. This undesired and uncontrolled exotherm is associated with the rapid generation of
cis- and
trans-2-butene as confirmed by
1H NMR analysis of the reaction mixture. These side processes caused significant increase in the reaction volume and a significant attenuation of the rate of the desired reaction. It was thus necessary to remove the ethylene line from the vessel and allow for the solution to cool. After the introduction of the substrate
6 and reintroduction of ethylene (maintained at a pressure of 1 atm), the exotherm and increase in volume were negligible. A low internal temperature was easily maintained and the desired reaction was facile and highly selective.
32.
Reagent grade
pentane was purchased from Fisher Scientific Co. and was used as received.
33.
The more polar, colored impurities were effectively removed.
34.
Based on
1H NMR analysis, reaction conversion was higher than 99% and compound
7 was the only constitutional isomer observed. The product thus obtained was of very high purity. Analytically pure sample was prepared by a fractional distillation of a portion of the product at 15 mmHg and collecting the fraction boiling at 82–83 °C.
35.
The product displayed the following physicochemical properties: [a]
D22 = -22.3 (
c 1.05, CHCl
3), lit.
10b [a]
D20 = -12.5 (
c 0.8, CHCl
3, 96:4 er);
1H NMR
pdf (500 MHz, CDCl
3) δ: 7.34-7.28 (m, 4 H), 7.21-7.16 (m, 1 H), 6.03 (dd,
Jtrans = 17.4 Hz,
Jcis = 10.4 Hz, 1 H), 5.10 (dd,
Jcis = 10.7 Hz,
Jgem = 1.2 Hz, 1 H), 5.04 (dd,
Jtrans = 17.6 Hz,
Jgem = 1.2 Hz, 1 H), 1.89-1.72 (ABX
3, ν
A = 1.84, ν
B = 1.77,
JAB = 13.7 Hz,
JAX = 7.4 Hz,
JBX = 7.4 Hz, 2 H), 1.35 (s, 3 H), 0.77 (t,
J = 7.4 Hz, 3 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 147.4, 146.9, 128.0, 126.7, 125.7, 111.7, 44.5, 33.4, 24.3, 8.9; IR (neat) cm
−1: 3083, 3058, 3023, 2968, 2935, 2878, 1944, 1873, 1830, 1801, 1635, 1600, 1493, 1456, 1445, 1379, 1370, 1030, 1003, 914, 760, 700; GC:
tR 10.61 min (poly(dimethylsiloxane), 25 m × 0.25 mm, 1.0 μm film thickness, 1.0 mL helium/min (1:100 split), 5 min at 100 °C, 5°C/min, 5 min at 200 °C); CSP-GC:
tR (
R)-
7, 24.28 min (98.8), (
S)-
7, 25.16 min (1.2). (Cyclodex B (J & W Scientific, 30 m × 0.25 mm, 0.25 μm film thickness) hydrogen (1.40 bar), 1:1 split, 30 min at 65 °C). Anal. Calcd. for C
12H
16: C, 89.94; H, 10.06. Found: C, 90.03; H, 10.16.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The procedure described here for the asymmetric hydrovinylation of 2-phenyl-1-butene (
6) with the generation of a quaternary carbon center
2 has also been used with minor modifications for related asymmetric hydrovinylations of various other vinylarenes, 1,3-dienes and norbornene. The product
7 has been prepared previously by (i) stoichiometric γ-alkylation of an allylic Grignard reagent using a methylating agent with a chiral leaving group (38% yield, 32% ee)
9 and, (ii) Cu-catalyzed asymmetric alkylation of an allylic phosphate with an organozinc reagent (94% yield, 97% ee).
10 Advantages of the present method
2,11 include the use of simpler starting materials (2-phenyl-1-butene and ethylene) and nearly salt-free reaction conditions, still retaining the high yield and enantioselectivity.
Among the Ni(II)-catalyzed olefin dimerization reactions, the hydrovinylation reaction, viz., the addition of a vinyl group and a hydrogen across a double bond (Scheme 1), has attracted the most attention.
12 Since the branched product is chiral, a regio- and stereoselective version of this reaction could provide easy access to a variety of olefin-derived products including carboxylic acid derivatives. For example, the hydrovinylation of vinylarene derivatives that leads to 3-arylbutenes could be used for the synthesis of widely used antiinflammatory 2-arylpropionic acids. Even though the hydrovinylation reaction has had a long history dating back to 1965, until recently no catalyst system gave satisfactory yield and selectivity to be of practical value. Most often the reaction is complicated by isomerization of the primary products, and oligomerization of the vinylarene and ethylene (Scheme 1). Besides, use of high pressures of ethylene and metal components incompatible with sensitive organic groups often limited the utility of many of the procedures reported earlier.
Scheme 1. Hydrovinylation of Vinylarenes
Since our disclosure
13 of new protocols for this reaction, its scope has been considerably broadened. Application of old (Figure 1,
8,
9,
12) and new (
10,
11,
13, 14) ligands have enabled successful asymmetric hydrovinylation of vinyl arenes,
14 1,3-dienes
14g,15 and strained bicyclic olefins such as norbornene.
14f,16 Several examples are illustrated in Eq. 1-6.
Figure 1. Assorted Ligands Used in Asymmetric Hydrovinylation Reactions
The search for new methods for stereoselective generation of quaternary carbon centers is a subject of considerable topical interest.
17 Several important pharmaceutically relevant compounds, among them, analgesic (-)-eptazocine,
18 protein kinase C activator lyngbyatoxin,
19 cognitive enhancing agent (-)-phenserine,
20 and serotonin antagonist LY426965,
21 contain an quaternary carbon center at the benzylic position. Hydrovinylation of 2-aryl-1-alkenes
13b generate a quaternary center at the benzylic position (Eq. 7) and introduces a highly versatile latent functionality in the form of a vinyl group. The resulting intermediates could be quite valuable for further synthetic elaboration. An asymmetric variant of this reaction (Eq. 8) forms the basis of this submission.
2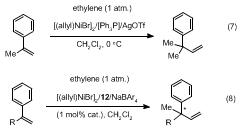
In scouting studies using 2-phenyl-1-butene (
6) as substrate, catalysts derived from the MOP ligand (Fig. 1,
9) show no reactivity while those derived from phospholane ligand
10, which gave high ee's and turnover numbers in the hydrovinylation of a number of styrene derivatives
14c and 1,3-dienes,
15 show only moderate reactivity under similar conditions.
2 Among the chiral ligands examined, the phosphoramidite
12 is found to provide the best results. Ligand
12, when treated with [(allyl)NiBr]
2 followed by NaBARF gave a very active pre-catalyst that effects the hydrovinylation of 1-ethylstyrene at −78 °C (4 h), with as little as 1 mol% of catalyst to give a nearly quantitative reaction.
2 Under these conditions, no isomerization or oligomerization products is detected, as judged by careful GC analysis and
1H NMR spectroscopy. The yields and selectivities are highly reproducible, and as expected, best selectivity is observed at low temperatures. They are independent of the catalyst loading or extent of reaction, clearly indicating the total absence of non-selective reactions.
Results of asymmetric hydrovinylation of several 2-aryl-1-alkenes under the optimal conditions are tabulated in Table 1. While the 4-methyl substrate
15 gave excellent selectivity for the formation of the expected product, the 4-chloro derivative
16 gave up to 5% isomerization of the starting olefin (entry 3). A similar minor side reaction was also observed for the substrates
18 and
20. An isopropyl group at the 1-position of the styrene (
17) retards the reaction (entry 4), and it is best accomplished at 24 °C with 10 mol% catalyst. Even though the yield of the reaction is only moderate, very high ee (~97%) was observed for the isolated product. The 2-naphthyl derivative
19 gave excellent yield (>98%) and selectivity (>99%) for the expected product. The tetralin derivative
20 represents a different class of substrates that underwent the hydrovinylation reaction giving >95% ee. Significant isomerization (~30%) of the starting material to an endocyclic olefin is a major distraction of this otherwise useful reaction. Compounds (e.g.,
21b) structurally related to the HV product
21a from
20 have been synthesized previously via intramolecular asymmetric Heck reactions (~93% ee),
18 stoichiometric oxazoline directed alkylation (~99% ee),
22a and enzyme-catalyzed desymmetrization of a chiral malonate (97% ee).
22b By comparison, the asymmetric hydrovinylation route is significantly shorter, and operationally simpler.
Table 1. Asymmetric Hydrovinylation of 2-Aryl-1-alkenesa
Among the other olefins 22-24, only the acyclic diene 24 undergoes hydrovinylation, and the product 25 is formed in nearly racemic form, contaminated with product of ethylene addition at the benzylic position.
Figure 2
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Benzophenone; (119-61-9)
Iodine; (7553-56-2)
3,5-Bis(trifluoromethyl)bromobenzene; (328-70-1)
Sodium tetrafluoroborate; (13755-29-8)
Bis[1,2:5,6-η-(1,5-cyclooctadiene)]nickel:
[bis(1,5-cyclooctadiene)nickel (0)]; (1295-35-8)
Allyl bromide; (106-95-6)
Ethylene; (74-85-1)
n-Butyllithium; (109-72-8)
Methyltriphenylphosphonium bromide; (1779-49-3)
Triphenylphosphine oxide; (791-28-6)
Sodium tetrakis[(3,5-trifluoromethyl)phenyl]borate; (79060-88-1)
Di(μ-bromo)bis(η-allyl)nickel(II):
[allylnickel bromide dimer]; (12012-90-7)
2-Phenyl-1-butene; (2039-93-2)
(R)-3-Methyl-3-phenylpentene:
[(1R)-1-ethyl-1-methyl-2-propenyl]-benzene]; (768392-48-9)
 |
T. V. (Babu) RajanBabu received his undergraduate education in India (Kerala University and IIT, Madras). He obtained a Ph.D. degree from The Ohio State University in 1978 under the direction of Professor Harold Shechter, and was a postdoctoral fellow at Harvard University with the late Professor R. B. Woodward. He then joined the Research Staff of Dupont Central Research, becoming a Research Fellow in 1993. He returned to OSU as a Professor of Chemistry in 1995. His research interests are in new practical methods for stereoselective synthesis focusing on enantioselective catalysis, free radical chemistry, and applications in natural product synthesis.
|
 |
Craig R. Smith was born in 1983 in Columbus, OH, USA. He graduated with his B.S. in Chemistry in 2003 and M.S. in Chemistry in 2005 from Youngstown State University under the direction of Professor Peter Norris. The same year, he started his Ph.D. studies at The Ohio State University under the supervision of Professor T. V. RajanBabu. His current interests are the development of asymmetric carbon-carbon, carbon-nitrogen and carbon-sulfur bond forming reactions, and the synthesis of biologically active heterocyclic natural products and analogs thereof.
|
 |
Aibin Zhang was born in China in 1974. After completing his B.S. in Chemistry at Anhui Normal University in 1996, he entered Shanghai Institute of Organic Chemistry, where he got his Ph.D. under the guidance of Dr. Shengming Ma in 2001. After a brief postdoctoral stint with Dr. Sean M. Kerwin at The University of Texas-Austin, he joined Professor RajanBabu's research group at The Ohio State University. His research at Ohio State focused on the development and application of asymmetric hydrovinylation reaction. Currently he is working for PharmaCore, NC.
|
 |
Dan Mans was born in St. Louis, MO and got his undergraduate education at St. Louis University in 2002. He joined Department of Chemistry at the Ohio State University for his Ph.D., which was completed in 2008 under the supervision of Professor RajanBabu. In his graduate work he was involved with the development of the hydrovinylation reaction and its applications for the synthesis of pseudopterosins and helioporins. His research interests are in development and optimization of organic reactions of broad applicability. In his current position in the pharmaceutical industry, he plans to use his synthetic skills in the area of medicinal chemistry.
|
 |
Min Xie received his bachelor's degree in Chemistry from Peking University in China. During that time he performed research with Professor Kai Wu on the fabrication of nanoporous anodic aluminium oxide (AAO) film and its application in the preparation of carbon nanotubes. In 2003 he began his graduate studies in Organic Chemistry at the University of Illinois, under the mentorship of Professor Scott E. Denmark. His current research focuses on the synthesis of rationally designed quaternary ammonium salts and investigation of phase transfer catalysis (PTC) by quantitative structure activity relationships (QSAR).
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved