Org. Synth. 2008, 85, 295
DOI: 10.15227/orgsyn.085.0295
(R)-2-(BENZYLOXYCARBONYLAMINO-METHYL)-3-PHENYLPROPANOIC ACID (Z-β2hPHE-OH)
[PREPARATION OF A β2-AMINO ACID WITH THE AUXILIARY DIOZ]
Submitted by J. Constanze D. Müller-Hartwieg
1, Luigi La Vecchia
1, Hartmut Meyer
2, Albert K. Beck
3, and Dieter Seebach
3.
Checked by Scott E. Denmark, Nathan Duncan-Gould, and Andrew Hoover.
1. Procedure
A. (4S)-4-Isopropyl-5,5-diphenyl-3-(3-phenylpropionyl)oxazolidin-2-one (1). In a 1-L, three-necked, round-bottomed flask equipped with an overhead mechanical stirrer (fitted with an 8-cm Teflon paddle), a Teflon-coated thermocouple, and a 150-mL pressure-equalizing addition funnel fitted with a nitrogen inlet, a solution of (4S)-4-isopropyl-5,5-diphenyloxazolidin-2-one (DIOZ) (Note 1) (22.5 g, 80 mmol) in tetrahydrofuran (THF) (266 mL) (Note 2) is cooled to −40 °C using an ethanol/dry ice bath. n-Butyllithium (1.6 M in hexane, 52.75 mL, 84.8 mmol, 1.06 equiv) (Note 3) is added to the well-stirred suspension (Note 4). The resulting reddish solution is allowed to warm to −15 °C (ca 15 min) and is then cooled to −70 °C. A solution of 3-phenylpropanoyl chloride (14.84 g, 87.97 mmol, 1.1 equiv) (Note 5) in THF (42.5 mL) is added via the addition funnel to the solution while the temperature is kept between −70 and −60 °C. Addition is completed after 20 min and stirring is continued for 30 min further (Note 6). The reaction mixture is allowed to warm to room temperature, quenched with half-saturated aq. NH4Cl solution (210 mL) and then is transferred to a 1000-mL separatory funnel where it is extracted twice with EtOAc (160 mL, 25 mL). The combined organic phases are washed consecutively with water (105 mL), sat. aq. NaHCO3 solution (21 mL), and half-saturated brine (105 mL). After being dried over MgSO4 (8 g, 30 min), the organic phase is filtered through an 8-cm medium-porosity glass filter funnel, then the filtrate is concentrated by rotary evaporation (23 °C, 15 mmHg) and then is dried at 0.2 mmHg for 8 h at room temperature. The crude product (35 g) is dissolved in refluxing tert-butyl methyl ether (TBME) (105 mL) and hexane (215 mL) is added slowly over 20 min. At the end of the addition, precipitation occurs. The mixture is then cooled in an ice bath for 2 h and is filtered through an 8-cm medium-porosity glass filter funnel to obtain the pure product, which is dried (23 °C at 0.08 mmHg) for 4 h to obtain 29.6 g (89%) of 1 as colorless needles (Note 7).
B. (2R,4S) [2-Benzyl-3-(4-isopropyl-2-oxo-5,5-diphenyl-3-oxazolidinyl)-3-oxopropyl]carbamic acid benzyl ester (2). A 1-L, three-necked, round-bottomed flask, fitted with a 150-mL, pressure-equalizing, addition funnel fitted with a nitrogen inlet, Teflon-coated thermocouple, a rubber septum, and a magnetic stirring bar (Note 8), is charged under a nitrogen atmosphere with 1 (28.9 g, 70.0 mmol) and dichloromethane (280 mL) (Note 9). The clear, colorless solution is cooled to −50 °C (ethanol/dry ice bath). A solution of TiCl4 in dichloromethane (1 M, 73.5 mL, 73.5 mmol, 1.05 equiv) (Note 10) is added dropwise via cannula keeping the temperature between -50 and −40 °C (Note 11). Then a 1 M solution of triethylamine in dichloromethane is added via syringe (ca. 3 mL) until the dark red color of the enolate persists (Note 12). More triethylamine (10.19 mL, 7.4 g, 73 mmol, 1.05 equiv) in dichloromethane (70 mL) is slowly added over 10 min keeping the temperature between −40 and −35 °C. After stirring the reaction mixture for 20 min further at this temperature, a solution of benzyl isopropoxymethyl carbamate (16.4 g, 73.5 mmol, 1.05 eq) (Note 13) in dichloromethane (80 mL) is added over 10 min via the addition funnel, followed by one more equivalent of the 1 M TiCl4 solution in dichloromethane (73.5 mL, 73.5 mmol, 1.05 equiv) via cannula keeping the temperature at −40 °C (Note 10). After 10 min, the cooling bath is replaced by an ice bath and the reaction mixture stirred for 3 h at 0–2 °C (Note 6). The yellow reaction mixture is quenched with a solution of sat. aq. NH4Cl solution (300 mL) and water (100 mL), then is stirred for 5 min and is transferred to a 1-L separatory funnel. The aqueous phase is separated and extracted with dichloromethane (50 mL). The combined organic layers are washed with half-saturated brine, dried over MgSO4 (15 g, 30 min), filtered through an 8-cm medium-porosity glass filter funnel, concentrated by rotary evaporation (23 °C, 15 mmHg) and dried in vacuo (23 °C, 0.08 mmHg) for 5 h to afford the crude product as a pale-yellow foam (42.7 g) (Note 14). The residue is dissolved, with stirring, in 250 mL of refluxing tert-butyl methyl ether (TBME). During the addition of hexane (150 mL) some seed crystals were added (Note 15, 16). The mixture is allowed to cool for 30 min and then is heated back to reflux. Another portion of hexane (100 mL) is added and the mixture is allowed to cool to room temperature overnight. The resulting crystals are collected by suction filtration through an 8-cm medium-porosity glass filter funnel, then are washed with two 100-mL portions of a mixture of ice-cold hexane/TBME, 2:1. After a second recrystallization from hexane/TBME, 1:1 (250 mL each), the isolated crystals are dried in vacuo (23 °C, 0.15 mmHg) for 8 h at room temperature to provide 21.05 g (52%) of the product 2 as colorless, diastereomerically pure microcrystals (Notes 17, 18).
C. (R)-2-(Benzyloxycarbonylaminomethyl)-3-phenylpropanoic acid (3). A 1-L, two-necked, round-bottomed flask fitted with an overhead stirrer (fitted with an 8-cm Teflon paddle), and a Teflon-coated thermocouple is charged with 2 (20.0 g, 34.9 mmol), THF (204 mL) and water (102 mL). Hydrogen peroxide (30%, 14.3 mL, 139.6 mmol, 4.0 equiv) (Note 19) is then added and the resulting mixture cooled to 0–5 °C in an ice bath. Lithium hydroxide monohydrate (2.34 g, 55.8 mmol, 1.6 equiv) (Note 20) is added and stirring is continued at 0–5 °C for 2 h (Notes 6, 21). The suspension is allowed to warm to room temperature, then is slowly diluted with aq. Na2SO3 solution (20 g in 160 mL, 158 mmol) (Note 22, 23) followed by 1 M aq. NaOH solution (102 mL) and stirring is continued at room temperature for 10 min. The THF is removed on a rotary evaporator (40 °C, 15 mmHg, 30 min) and TBME (635 mL) is added to the resulting suspension. After the mixture is stirred for 10 min, the precipitate (recovered DIOZ auxiliary) is filtered off by suction through an 8-cm medium-porosity glass filter funnel and is successively washed with 1 M aq. NaOH solution (101 mL), water (2 × 83 mL) and TBME (2 × 83 mL) (Note 24). The filtrate and all aqueous washes are combined and acidified to pH 2-3 with 0.52 M aq. citric acid solution (Note 25) and then are transferred to a 4-L separatory funnel and are extracted with TBME (2 × 255 mL). The combined TBME phases are washed with brine (255 mL), dried over MgSO4 (19 g, 10 min), filtered through an 8-cm medium-porosity glass filter funnel and concentrated by rotary evaporation (45 °C, 15 mmHg). The resulting oil slowly solidifies upon standing (Note 26) to afford 10.70 g (98%) of 3 with an enantiomer ratio of = 99.9:0.1 (Notes 27, 28, 29).
2. Notes
1.
This auxiliary (DIOZ) was prepared following the
Organic Syntheses procedure, ref. 4. Use of the corresponding
R-auxiliary leads to the
S- β
2 amino acid derivative
ent-
3.
2.
THF was purchased from Fisher Scientific and was dried by percolation through 4Å molecular sieves immediately prior to use.
3.
n-Butyllithium in hexane was purchased from Fluka and was titrated by the method of Gilman (1.62 M).
4.
The addition is slightly exothermic, therefore slow addition is recommended.
5.
3-Phenylpropanoyl chloride (98%) was purchased from ACROS and was used as received.
6.
The progress of the reactions can be monitored by TLC analysis (visualization with UV, I
2 and KMnO
4). Step A: (silica gel, hexane/EtOAc, 4:1); DIOZ: R
f = 0.15,
1: R
f = 0.75. Step B: (silica gel, hexane/EtOAc, 4:1);
1: R
f = 0.75,
2: R
f =
0.3. Step C: (silica gel, hexane/EtOAc, 3:2);
2: R
f = 0.85,
3: R
f = 0.11.
7.
The product exhibits the following physicochemical properties: mp 98–99 °C; [α]
D23 = -176.2 (c 1, CHCl
3);
1H NMR
pdf (400 MHz, DMSO) δ: 0.6, 0.85 (2 d,
J = 7.1, 6.87 Hz, 6 H, 2 × CH
3), 2.03 (dp,
J =7.1, 2.5 Hz, 1 H, CH), 2.75 (t,
J = 7.9 Hz, 2 H, CH
2-phenyl), 2.96, 3.14 (2 m, 2 H, CH
2-CO), 5.59 (d
, J = 2.6 Hz, 1 H, CHN), 7.05-7.65 (m, 15 H, aryl-H).
13C NMR
pdf (125 MHz, DMSO) δ: 171.3, 152.6, 143.1, 140.2, 138.2, 128.8, 128.38, 128.30, 128.2, 128.1, 127.7, 125.9, 125.3, 124.9, 88.7, 64.4, 36.2, 29.8, 29.6, 21.2, 15.4; IR (KBr) cm
−1: 2961 (m), 2938 (m), 1781 (s), 1700 (s); MS (EI)
m/z (relative intensity, %): 413 (41), 263 (16), 222 (43), 207 (41), 183 (46), 165 (28), 133 (26), 117 (58), 105 (100). Anal. Calc. for C
27H
27NO
3: C 78.42, H 6.58, N 3.39. Found: C, 78.53; H, 6.69; N, 3.54.
8.
A 5-cm Rugby-ball-shaped stir bar was used.
9.
Dichloromethane purchased from Fisher Scientific and was dried by percolation through 4 Å molecular sieves immediately prior to use.
10.
Titanium tetrachloride (1 M in CH2Cl2) was purchased from Fluka and was used as received.
11.
Although the submitters added the solution
via syringe, on this scale the checkers found it more convenient to transfer the titanium tetrachloride solution
via cannula and a 100-mL graduated cylinder (flame dried, with septa and a nitrogen needle inlet.
12.
Triethylamine (Reagent Grade) was purchased from Aldrich Chemical Co. and was freshly distilled from CaH
2 prior to use.
13.
For the preparation of this electrophile see the preceding procedure in this volume.
14.
The crude product contains the desired (
R,S)-diastereomer, as well as the (
S,
S)-epimer in a 9:1 ratio. As a by-product (
ca. 7%) 2-benzyl-3-(4-isopropyl-2-oxo-5,5-diphenyloxazolidin-3-oxopropyl)carbamic acid isopropyl ester is formed. Both by-products can be removed by recrystallization.
15.
Seed crystals can be obtained after addition of hexane to a TBME solution of a small amount of crude product.
16.
The checkers found that a hot filtration of the TBME solution through
~1.5 g of Celite (acid-washed) in a cotton-plugged funnel removed a small amount (~5%) of triethylamine hydrochloride and made the recrystallization easier.
17.
An additional 12% of product
2 can be obtained by chromatography of the mother liquors (column dimensions: 12 cm × 6 cm (h × d), 200 g of SiO
2, hexane/EtOAc, 85:15 (1 L), 80:20 (500 mL), 75:25 (500 mL), 70:30 (500 mL), 65:35 (500 mL) and subsequent crystallization (TBME/hexane). The mother liquors from this crystallization contain a 1:1 mixture of diastereomers that aids in the assignment of the products (see
Note 13). Apart from the product, 19% of starting material can be recovered by this chromatography.
18.
The product exhibits the following physicochemical properties: mp 129.5–130.5 °C; [α]
D23 -96.2 (c 1, CHCl
3);
1H NMR
pdf (500 MHz, DMSO) δ: 0.6, 0.85 (2 d,
J = 7.0 Hz, 6 H, 2 × CH
3), 2.03 (m, 1 H, CH-
i-Pr), 2.42, 2.59 (2 × dd, 2 H,
J = 14.1, 6.5 Hz, CH-C
H2-phenyl), 3.23 (m, 2 H, CH
2), 4.11 (m, 1 H, CH), 4.98 (dd, 2 H,
J = 21.7, 12.7 Hz, O-CH
2-phenyl), 5.54 (d, 1 H,
J = 2 Hz, CHN), 6.65-7.63 (m, 21 H, aryl-H + NH).
13C NMR
pdf (125 MHz, CDCl
3) δ: 173.0, 126.8, 153.1, 143.8, 138.84, 138.81, 138.29, 137.8, 137.8, 129.5, 129.2, 129.1, 129.0, 128.97, 128.7, 128.45, 128.40, 128.3, 126.7, 126.0, 125.4, 89.2, 65.9, 65.6, 44.7, 42.0, 35.1, 30.2, 21.8, 16.0. IR (KBr) cm
−1: 3366 (s), 3064 (m), 3029 (m), 2870 (m), 1775 (s), 1716 (s), 1704 (s), 1539 (s) MS (EI)
m/z (relative intensity,%): 576 (4), 412 (45), 222 (16), 167 (15), 131 (28), 91 (100). Anal. Calc. for C
36H
36N
2O
5: C 74.98, H 6.29, N 4.86. Found: C 75.02, H 6.32, N 4.94.
19.
Hydrogen peroxide (Trace Select ≥ 30%) was purchased from Fluka and was used as received.
20.
Lithium hydroxide (Purum p.a. ≥ 99%) was purchased from Fluka and was used as received.
21.
This procedure was used as described in ref. 6 and 7.
22.
The temperature was maintained at 18–24 °C by re-immersion in the ice bath.
23.
Sodium sulfite (Reagent Grade, 98%) was purchased from Sigma-Aldrich Co. and was used as received.
24.
The recovered DIOZ-auxiliary (9.34 g, 95%, >95% purity by
1H NMR) can be reused.
25.
Citric acid (99%) was purchased from the Aldrich Chemical Co. and was used as received.
26.
The checkers found that rotary evaporation (45 °C, 15 mmHg) followed by high vacuum (0.2 mmHg, 12 h) was sufficient to obtain a solid. Suspending this solid in 150 mL of TBME followed by filtration and further rinsing with
TBME (2 × 20 mL) generated analytically pure material after removing the solvent (45 °C, 15 mmHg, 3 h; then 0.2 mmHg, 23 °C, 30 h).
27.
The enantiomer ratio
er was determined by HPLC:
tR (
S)-
3 = 23.33 min (<0.1%), (
R)-
3 = 27.69min (>99.9%) (Chiralpak AD-H; hexane/EtOH/MeOH, 92:4:4 + 0.1% TFA; 1 mL/min, 10 μg in 10 μL of MeOH, UV 210 nm.
28.
If sodium hydroxide is used instead of LiOH/H
2O
2 (as described in ref. 5) partial racemization was observed, and the er dropped to 90:10.
29.
The product exhibits the following physicochemical properties: mp 73.0–75.0 °C; [α]
D23 = -4.1 (c 1, EtOH);
1H NMR
pdf (500 MHz, DMSO) δ: 2.77 (m, 3 H, CH-CH
2-phenyl), 3.15, 3.22, (2 m, 2 H, CH
2), 5.02 (s, 2 H, O-CH
2-phenyl), 7.15-7.46 (m, 11 H, aryl-H + NH), 12.3 (bs, 1 H, -CO
2H);
13C NMR
pdf (125 MHz, DMSO) δ: 174.8, 156.3, 139.2, 137.3, 128.9, 128.4, 128.3, 127.84, 127.75 126.2, 65.3, 47.2, 42.2, 35.2; IR (KBr) cm
−1: 3346 (br, s), 3031 (br, s), 2662 (m), 1700 (s); MS
m/
z (relative intensity, %): 313 (2.9), 149 (38), 131 (23), 117 (29), 108 (29), 107 (27), 103 (11), 92 (20), 91 (100), 79 (31), 78 (10), 77 (27), 74 (10), 65 (23). Anal. Calc. for C
18H
19NO
4: C 68.99, H 6.11, N 4.47. Found: C 68.93, H 6.75, N 4.62.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The procedure described herein is an application of the Evans oxazolidinone-auxiliary method to the overall enantioselective derivatization of an achiral carboxylic acid with electrophiles (
4 →
5).
8 Instead of one of the classical auxiliaries the valine-derived 4-isopropyl-5,5-diphenyl-oxazolidinone (DIOZ) is used, which has many practical advantages outlined in previous publications of our group
6,9 and in an
Organic Syntheses procedure for its preparation.
4 The transformation of the present procedure is a carbamidomethylation (
Mannich reaction) of 3-phenylpropionic acid (R = CH
2, R
E = CH
2NHCO
2Bn in
4,
5). The actual electrophile (Bn-O-CO-
+NH=CH
2), generated
in situ by the action of the Lewis acid TiCl
4 on benzyl isopropoxymethyl carbamate (Bn-O-CO-NH-CH
2-O-CHMe
2), reacts with the Ti-enolate of the
N-(3-phenylpropionyl)oxazolidinone
1 to give β-amino-acid derivative
2 with a diastereoselectivity of 9:1. Starting material
1, the minor stereoisomer
epi-
2 and other by-products can be readily removed by crystallization when the benzyl
isopropoxymethyl carbamate is employed, rather than the
methoxymethyl carbamate, which was originally used
10 for this type of reaction
(Note 13).
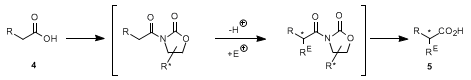
The product, described herein, for preparing β
2-amino-acid derivatives is of general importance: it has been applied to the following building blocks for β-peptide synthesis, with Fmoc protecting group PG and, where applicable, with appropriate acid-labile protection in the side chain R: β
2hAla, β
2hPhe, β
2hTyr, β
2hVal, β
2hLeu, β
2hIle, β
2hMet, β
2hArg, β
2hPro, β
2hLys.
5,6,11,12,13 Actually, all β
2-amino acids with proteinogenic side chains have been obtained with the help of the auxiliary DIOZ.
13 For numerous other methods of preparing certain β
2-amino acids, including derivatives of β
2hPhe, such as compound
3, we refer the reader to the literature.
13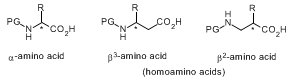
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(4S)-4-Isopropyl-5,5-diphenyloxazolidin-2-one (DIOZ):
2-Oxazolidinone, 4-(1-methylethyl)-5,5-diphenyl-, (4S)-; (184346-45-0)
n-Butyllithium:
Butyllithium; (109-72-8)
3-Phenylpropanoyl chloride:
Benzenepropanoyl chloride; (645-45-4)
(4S)-4-Isopropyl-5,5-diphenyl-3-(3-phenyl-propionyl)oxazolidin-2-one:
2-Oxazolidinone, 4-(1-methylethyl)-3-(1-oxo-3-phenylpropyl)-5,5-diphenyl-, (4S)-; (213887-81-1)
Titanium tetrachloride; (7550-45-0)
Triethylamine:
Ethanamine, N,N-diethyl-; (121-44-8)
(2R,4S) [2-Benzyl-3-(4-isopropyl-2-oxo-5,5-diphenyl-3-oxazolidinyl)-3-oxopropyl]carbamic acid benzyl ester:
Carbamic acid, [(2R)-3-[(4S)-4-(1-methylethyl)-2-oxo-5,5-diphenyl-3-oxazolidinyl]-3-oxo-2-(phenylmethyl)propyl]-, phenylmethyl ester; (218800-56-7)
Hydrogen peroxide; (7722-84-1)
Lithium hydroxide monohydrate; (1310-66-3)
(R)-2-(Benzyloxycarbonylaminomethyl)-3-phenylpropanoic acid:
Benzenepropanoic acid, α-[[[(phenylmethoxy)carbonyl]amino]methyl]-, (αR)-; (132696-47-0)
 |
Dieter Seebach received his Ph.D. from the Technische Hochschule Karlsruhe, Germany, with Rudolf Criegee. After a postdoctoral stay in Elias J. Corey's group and a lectureship at Harvard University, he returned to the University of Karlsruhe and became a full Professor of Organic Chemistry at the Justus-Liebig-Universität in Giessen, Germany, in 1971. From 1977-2003 he was Professor of Chemistry at the Eidgenössische Technische Hochschule (ETH) in Zürich. Since 2003 he is officially retired; as Professor Emeritus he continues doing research with postdoctoral coworkers. His research activities include: development of new synthetic methods, natural-product synthesis, structure determination, chiral dendrimers, the biopolymer PHB, β-peptides. The results have been described in over 800 research papers.
|
 |
J. Constanze D. Müller-Hartwieg studied chemistry at the University of Konstanz, Germany. After her Ph.D. at the Max-Planck-Institute of Biochemistry in Martinsried/Munich, Germany, in the field of bioorganic chemistry and peptide synthesis, she joined Novartis in Basel, Switzerland as a Postdoc working in the Combinatorial Chemistry Unit. Since 2002 she has been working as a Lab Head in the Preparation Laboratories for Novartis Institutes for BioMedical Research, Basel. She is responsible for the scale up of organic syntheses, in order to produce larger amounts of building blocks, intermediates and potential drug candidates for a wide range of disease areas.
|
 |
Luigi La Vecchia did his Ph.D. 1989 at the University of Saarbrücken, Germany. Thereafter, he joined Sandoz as Lab Head of the Research Kilo lab. Between 1993 -1994 he did a sabbatical at the Sandoz Research Institute/Chemical Development in East Hanover, NJ. In 1996 he took over a position as Lab Head Process R&D in Chemical Development in Basel, Switzerland. Since 1998 he has been assuming various positions within the Novartis Institute for Biomedical Research, Basel, Switzerland. Currently, he is Director of the Preparations Laboratories. He was involved in projects such as RAD001 (Certican), QAB149 (Indacaterol) and NIM811.
|
 |
Hartmut H. Meyer graduated in 1970 from the Technische Hochschule Hannover (Germany) under the supervision of W. Theilacker and F. Klages. After postdoctoral work with Professor D. Seebach at the University of Giessen in 1972, he returned to the University of Hannover and started work on syntheses of enantiopure natural products. In 1987 he became Privatdozent with a Habilitation on studies of the benzidine rearrangement. Since 1996 he has been a Professor at the Leibniz Universität Hannover. His current research interests are synthetic methodology, chemo-enzymatic syntheses and biochemical studies, in cooperation with other research groups.
|
 |
Albert Karl Beck was born in 1947 in Karlsruhe (Germany), completing a chemistry technician's apprenticeship at the University of Karlsruhe in 1966, and joined the Seebach research group in 1968. Between 1969 and 1972 he continued his education, obtaining official certification as a chemical technician at the Fachschule für Chemotechnik in Karlsruhe. In 1971 he joined the Institute for Organic Chemistry at the University of Giessen, and in 1974 he engaged in a six-month research visit to the California Institute of Technology in Pasadena (USA). He has been an active part of the Laboratory for Organic Chemistry at the ETH in Zürich since 1977. During his association with the Seebach research group he has participated in essentially all of the groups research themes, as evidenced by his coauthorship of more than 80 publications.
|
 |
Nathan W. Duncan-Gould received his bachelor's degrees in Molecular Biology, Biochemistry and Chemistry from the College of Charleston. During this time he performed research with Professor Justin K. Wyatt on the total synthesis of Cytosporone E. In 2004 he began his graduate studies in Organic Chemistry at the University of Illinois, unde the mentorship of Professor Scott E. Denmark. The focus of his research is the investigation of phase transfer catalysis (PTC) by quantitative structure activity relationships (QSAR).
|
 |
Andrew J. Hoover was born in New London, Connecticut, in 1987. He enrolled at the University of Illinois in 2005, and beginning in 2006 he conducted research on deoxyribozyme catalysis with Professor Scott Silverman. As an Amgen scholar during the summer of 2007, he worked with Professor Richmond Sarpong at UC Berkeley on efficient syntheses of cyclopentenones, and in the fall of 2007 he joined the research group of Scott Denmark, where he currently is investigating the use of Lewis bases as catalysts for enantioselective functionalization of alkenes. He will graduate in December 2008 with a chemistry BS degree, and will enroll in graduate school in 2009.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved