Org. Synth. 2009, 86, 172
DOI: 10.15227/orgsyn.086.0172
CONVENIENT PREPARATION OF 3-ETHOXYCARBONYL BENZOFURANS FROM SALICYLALDEHYDES AND ETHYL DIAZOACETATE
Submitted by Matthew E. Dudley, M. Monzur Morshed, and M. Mahmun Hossain
1.
Checked by David Hughes
1. Procedure
Caution: Ethyl diazoacetate (EDA) and other diazo compounds are potentially explosive and therefore must be handled with caution. They are also toxic and prone to cause development of specific sensitivity. A well-ventilated hood should be used for the entire procedure.
5-Chloro-3-ethoxycarbonylbenzofuran. A 250-mL, three-necked, round-bottomed flask is fitted with a reflux condenser, thermometer, and a pennyhead stopper (Note 1). A 3-cm oval stir bar is added to the flask. 5-Chlorosalicylaldehyde (5.00 g, 31.9 mmol) (Note 2) is added to the flask along with 11 mL CH2Cl2 (Note 3) to form a thick white slurry, then 54% HBF4·OEt2 (0.50 g, 3.2 mmol, 0.1 equiv) (Note 4) is added by syringe directly to the stirring solution through the neck containing the pennyhead stopper, whereby a color change from a white slurry to an orange slurry is noted. The pennyhead stopper is then replaced with a clean and dry 100-mL pressure-equalizing addition funnel with the Teflon stopcock closed (Note 5). Ethyl diazoacetate (90% pure) (5.79 g, 45.7 mmol, 1.43 equiv) is added to the addition funnel and diluted to 33 mL with dichloromethane (Notes 5-7). The EDA solution is then added to the reaction mixture at a rate of 5-7 mL per minute, resulting in steady evolution of nitrogen (total addition time was 5-7 minutes) and an increase in temperature from the initial 23 °C to 38 °C with a gentle reflux (Notes 5, 6 and 8). Once addition is complete gas evolution ceases within a minute and the reaction is assayed for completion by 1H NMR (Note 9). A heating mantle is placed under the round-bottomed flask. The equalizing addition funnel is replaced with a pennyhead stopper. At this point the solution is dark yellow in color. A simple distillation apparatus is assembled in place of the original reflux condenser using a distillation adapter, Claisen head adapter, and a 100-mL receiving flask. The reaction mixture is concentrated by distilling CH2Cl2 (30-33 mL) and free EDA to a maximum pot temperature of 58-61 °C leaving a clear yellow viscous oil. After removal of the heating mantle the solution is cooled to room temperature using a water bath, then 4.0 g of 95-98% H2SO4 (39.2 mmol) (Note 10) is added by syringe directly to the stirring viscous oil through the neck containing the pennyhead stopper over a period of 1-2 minutes. The addition is only slightly exothermic, with a temperature rise of about 5 °C using a water-cooling bath, and a color change from a clear yellow oil to a cloudy yellow-orange suspension is noted. After 10 minutes, the second step is complete and ready for neutralization. With water bath cooling and rapid agitation, the acidic reaction mixture is neutralized by slow addition of 100 mL saturated NaHCO3 solution (Note 11). The empty addition funnel is removed and replaced with a pennyhead stopper. The water bath is removed and replaced with a heating mantle. The aqueous mixture is azeotropically distilled, while maintaining high speed stirring in order to remove any trace of residual CH2Cl2 to a maximum pot temperature of 90 °C. Upon reaching 90 °C, the heating mantle is removed and the aqueous mixture is allowed to cool to room temperature while continuing to maintain rapid stirring (Note 12). After crystallizing overnight, the crude yellow solid product is filtered using a Büchner funnel and dried to constant weight. The yield of crude material (~90% pure by NMR) is 5.99-6.08 g (84-86% uncorrected for purity).
The crude material is purified by recrystallization. To a 250-mL, three-necked, round-bottomed flask equipped with a thermometer, stopper, and 100 mL addition funnel are added a 3-cm oval stir bar, 30 mL of anhydrous EtOH, in which 0.3 g of NaOH has been dissolved, and the crude product (5.97 g) (Note 13). The mixture is stirred with warming to 35 °C in order to dissolve the product, then 70 mL of deionized water is added dropwise from the addition funnel over 15 min. The product begins to crystallize after 10 mL water is added. On completion of the water addition, the mixture is stirred for 5 min at room temperature, then filtered using a Büchner funnel and filter paper. The flask is washed with 2 × 15 mL of deionized water and the washes are then used to rinse the crystalline product on the Büchner funnel. The crystalline product is allowed to dry on the Büchner funnel for 30 min to 1 h to provide 4.84 g (68%) of product as a tan, free-flowing solid (Note 14).
2. Notes
1.
A mechanical stirrer and a four-necked flask can be substituted for the magnetic stir bar and three-necked flask apparatus with round-bottomed flasks smaller than 500 mL. The submitters strongly suggest use of a mechanical stirrer over magnetic stirring in order to maintain vigorous stirring and agitation as noted throughout the procedure.
2.
5-Chlorosalicylaldehyde (98%) was purchased from Alfa Aesar and was used as received. The checkers used 5-chlorosalicylaldehyde purchased from Aldrich. NMR suggested a purity of about 96%. Yields are uncorrected for purity.
3.
CH2Cl2 (99.5%) was purchased from VWR International and distilled from P
2O
5. The checkers used
dichloromethane (99.5%) from Sigma-Aldrich and used without purification. The water content was measured by Karl-Fischer titration and found to be <50 ppm.
4.
HBF4·OEt2 (tetrafluoroboric acid, 54% w/w in diethyl ether) solution was purchased from Aldrich Chemical Company as a clear colorless or clear yellow solution. Solutions that are dark brown or purple in color are unacceptable. The checkers added tetrafluoroboric acid based on weight; the tetrafluoroboric acid was drawn into a weighed syringe to the appropriate weight, discharged into the flask, and re-weighed to determine the accurate amount added.
5.
A Teflon stopcock is recommended for ethyl diazoacetate (EDA) as greased glass stopcocks tend to leak in the presence of the EDA solution. For reactions run with magnetic stirring, the addition funnel was placed on the middle neck such that the EDA could drop directly into the vortex of the stirring solution. The checkers used ethyl diazoacetate purchased from Aldrich. The EDA was assayed via NMR using toluene as an internal standard, as follows. Approximately 50 mg each of EDA and
toluene (99.5% purity) were accurately weighed into a 5-mm NMR tube, diluted with about 0.8 mL of CDCl
3, and analyzed using
1H NMR with a 5 second delay to ensure full relaxation. The CH
3 protons were integrated and compared to the integration of the CH
2 and CH
3 groups of EDA. A fresh bottle from Aldrich was assayed at 90 wt% with 9% dichloromethane. An older bottle assayed at 78 wt % with 15 wt% dichloromethane.
6.
The Merck Index cautions against using EDA with sulfuric acid.
2 In our hands, we have had no issues or incidents regarding use of tetrafluoroboric acid and EDA. Information is available concerning the safety of EDA on an industrial scale.
37.
The submitters report that the previously published preparation of EDA
4 can also be used. By omitting the final distillation of dichloromethane, the requisite 7-10 mol% EDA solution is obtained.
8.
In cases of larger scale reactions, overwhelming of the condenser by refluxing CH
2Cl
2 may be prevented by using a room temperature water bath to control excessive temperatures. The water bath is unnecessary for a 5 g scale reaction. The EDA solution should be dripped directly into the rapidly stirring reaction mixture and not on the walls of the flask.
9.
The completion of reaction was determined by
1H NMR. A 50 mg sample of the batch was diluted with 0.7 mL CDCl
3 and analyzed by NMR. The phenol and aldehyde resonances (at 10.9 ppm and 9.9 ppm) were integrated and compared to the combined integration of all resonances from 6 to 8 ppm (3 aromatic protons for starting material, intermediates, and products). If more than 5% unreacted aldehyde remained, an appropriate amount of EDA was added to complete the reaction.
10.
ACS reagent grade (95.0-98.0%) H
2SO
4 was purchased from Fischer Chemical Company and was used as received. The checkers added sulfuric acid by weight; the sulfuric acid was drawn into a weighed syringe to the appropriate weight, discharged into the flask, and re-weighed to determine the accurate amount added.
11.
ACS reagent grade NaHCO
3 (99.7-100 %) was purchased from Aldrich Chemical Company and was used as received. The initial addition of saturated bicarbonate should be dropwise due to a rapid exotherm; after the first 5 mL are added, the exotherm subsides as the mixture is cooled by CO
2 evolution. The final 95 mL can be added over 5 min.
12.
The entire azeotrope procedure lasts approximately 15 min and generally less than 1 mL is distilled. The majority of the CH
2Cl
2 distills at about 72 °C and the azeotrope is stopped once water is seen distilling into the receiver (90 °C). Upon cooling to room temperature the pH of the aqueous mixture was 8-9 as measured with pH paper. This part of the procedure is a trituration and requires rapid stirrer speed. The oily melt must be efficiently dispersed throughout the aqueous solution on cooling to room temperature or it will oil out and eventually form an impure gummy solid. In addition, the mixture must slowly cool to room temperature by equilibrating with the surrounding air. Forced cooling (i.e., ice baths, water jackets, etc.) should be avoided. In many cases, it may take several hours for complete trituration and eventual crystal formation to occur. It is suggested to let this process go overnight under the highest possible stir speed. The checkers found that if gumming occurred, the mixture could be re-heated to 80 °C and allowed to re-cool. This provided solids in all cases. As an alternate to crystallization from the aqueous solution, the checkers also used the following extractive procedure. The final distillation at 90 °C is omitted, and the aqueous mixture after neutralization is extracted with 2 × 50 mL ethyl acetate. The EtOAc extracts are combined, washed with 40 mL brine, filtered through a bed of sodium sulfate, and concentrated by rotary evaporation (30 mmHg, bath temp 40 °C). The resulting material crystallized on standing and was recrystallized from EtOH/water as described in the text to provide 5.26 g of product (74% yield).
13.
NaOH pellets were dissolved in ethanol using sonication. The recrystallization process can be repeated if needed for desired purity, although loss of yield is approximately 15% for each recrystallization. ACS reagent grade ethanol, absolute 200 proof (≥99.5%) and ACS reagent grade NaOH pellets, (≥97.0%) were used as received from Aldrich chemical.
14.
Physical and spectroscopic properties of 5-chloro-3-ethoxycarbonylbenzofuran are as follows: mp 60.5-61.5 °C;
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.43 (t,
J = 7.2 Hz, 3 H), 4.42 (q,
J = 7.2 Hz, 2 H), 7.32 (dd,
J = 2.1, 8.8 Hz, 1 H), 7.44 (d,
J = 8.8 Hz, 1 H), 8.03 (d,
J = 2.1 Hz, 1 H), 8.26 (s, 1 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 14.4, 60.8, 112.7, 114.6, 121.8, 125.6, 126.0, 130.0, 152.0, 153.9, 162.9; HRMS (EI)
m/z calcd for C
11H
9O
3Cl: 224.02407; found: 224.0241. Anal. Calcd for C
11H
9O
3Cl: C, 58.82; H, 4.04. Found: C, 58.54; H, 4.01.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
3-Substituted benzofuran derivatives are medicinally and biologically important compounds.
5 Many of the benzofuran syntheses to date result in the formation of 2-substituted or 2,3-disubstituted benzofurans, although 3-substituted benzofurans are not often reported. Nonetheless, at least three other synthetic methods have appeared recently. All three methods involve multi-step palladium-mediated intramolecular cyclizations from
o-halo phenols or
o-dihalobenzenes, which have limited commercial availability and high cost. One method involves carbonylative cyclization of complex
o-alkynylphenols.
6a,b,c A second involves cyclization by enolate
O-arylation.
6d A third method, recently reported by Malona, et al involves an improved synthesis of 3-ethoxycarbonyl benzofuran (entry 1) in 74% overall yield from iodophenol. The two-step method uses a Michael addition to ethyl propiolate to give
E-3-(2-iodophenoxy)-2-propenoic acid ethyl ester in 88% yield followed by an intramolecular Heck reaction (84% yield).
6e The synthetic procedure given here is higher yielding than the other three aforementioned synthetic methods. Furthermore, this procedure uses the relatively inexpensive and commercially available salicylaldehydes, in lieu of the more expensive
o-halo phenols or
o-dihalobenzenes.
7 The reaction has been shown to proceed through a unique hemiacetal which subsequently dehydrates to the benzofuran product as detailed in Scheme 1.
7 Upon protonation of the carbonyl oxygen, the activated salicylaldehyde undergoes nucleophilic attack by EDA, followed by an aryl migration with concomitant loss of N
2.
8 The resulting aryl propanal tautomerizes rapidly to form a 3-hydroxyaryl acrylate, which again isomerizes to a stable cyclic hemiacetal in the presence of acid.
7Scheme 1
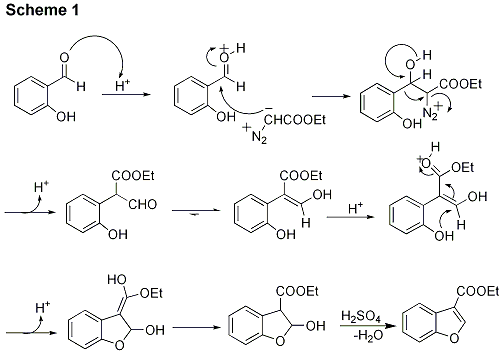
The following procedure can be generalized for any of the 3-substituted benzofurans represented in Table 1. No inert N2 atmosphere is required. The entire two-step, one-pot reaction can be accomplished in less than 2 hours. The procedure yields product of analytical purity after recrystallization. In cases where EDA is undertitrated, an excess of 5-chlorosalicylaldehyde starting material will remain. Conversely, overtitration of EDA results in formation of diethyl diglycolate impurity. Both of these impurities are easily observed by 1H NMR. The 1H NMR chemical shifts for the phenolic and aldehydic protons of 5-chlorosalicylaldehyde are: (CDCl3, 300 MHz) δ: 11.01 (s, 1 H), 9.83 (s, 1H) respectively. The 1H NMR chemical shifts for the methylene and -CH2 ester protons of diethyl diglycolate are: (CDCl3, 300 MHz) δ: 4.24 (s, 4H), 4.23 (q, 4H) respectively. Both 5-chlorosalicylaldehyde and diethyl diglycolate are acidic compared to the product and can therefore be removed by employing a recrystallization procedure from ethanolic NaOH solution.
Table 1. Isolated Yields of 3-Ethoxycarbonyl Benzofurans
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
3-Benzofurancarboxylic acid, 5-chloro-, ethyl ester; (899795-65-4)
EDA: Acetic acid, 2-diazo-, ethyl ester; (623-73-4)
Tetrafluoroboric acid diethyl etherate; (67969-82-8)
5-Chlorosalicylaldehyde: Benzaldehyde, 5-chloro-2-hydroxy-; (635-93-8)
 |
M. Mahmun Hossain received his M.Sc. degree in chemistry from Dhaka University, Bangladesh. In 1985, he received his Ph.D. from the University of South Carolina. After about 3 years of postdoctoral study with Professor Jack Halpern at the University of Chicago, he joined the Department of Chemistry at the University of Wisconsin-Milwaukee as an Assistant Professor. In 1994, he was promoted to Associate Professor, and received a research award from the UWM Foundation for his outstanding research and creativity. He is an author of nearly 60 publications and has presented more than 100 seminars, posters, and papers at various meetings, universities, and colleges. |
 |
Matt Dudley received a B.A. degree in German in 1994 from the University of Denver and an ACS-approved B.S. degree in both Chemistry and Biology in 1998 from the University of Wisconsin-Oshkosh. After an internship award at Aldrich Chemical Company, he joined Aldrich in 1998 as an Associate Chemist and was promoted to Chemist in 1999, where he received three awards for large scale and cGMP processes, and safety. He joined the Hossain group at the University of Wisconsin-Milwaukee in 2002 under fellowship. He received the 2006 Outstanding Teaching Assistant Award and is studying benzofurans and asymmetric-aryl quaternary carbon centers. |
 |
Monzur Morshed received his B.Sc. (Honors) and M.Sc. in Chemistry from Jahangirnagar University, Bangladesh. He also completed an M.S. degree in Wood Science from Mississippi State University (MSU). He began Ph.D. studies in Organic Chemistry at the University of Wisconsin-Milwaukee in 2002. Since then he has been an active member in Professor Mahmun Hossain's group and has received multiple Chancellor's Fellowship Awards. He was awarded an ACS Milwaukee Section Travel Grant in 2007. He was also recently honored with the University's Outstanding Teaching Assistant Award for 2007. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved