Org. Synth. 2023, 100, 61-83
DOI: 10.15227/orgsyn.100.0061
Preparation of N-Boc-3-methylpyrrole via Anionic Rearrangement
Submitted by Daniel P. Graeff, Noah J. Sims, Quentin R. Savage, Richard K. Jackson, III, Amy C. Jackson, Danielle M. Lawson, Madelyn R. Wixom, and John L. Wood*
1Checked by Yuya Shiga, Haruka Fujino, and Masayuki Inoue
1. Procedure (Note 1)
A. tert-Butyl 5-methyl-3,6-dihydro-2H-1,2-oxazine-2-carboxylate and tert-butyl 4-methyl-3,6-dihydro-2H-1,2-oxazine-2-carboxylate (2). A single-necked, 2 L, 29/32 round-bottomed flask is charged with a 4-cm by 1.5-cm Teflon-coated football-shaped stir bar and charged with methylene chloride (750 mL) (Note 2) under air, measured using a 1 L graduated cylinder and introduced into the reaction vessel through a 350 mL plastic funnel. The funnel is removed from the flask. Stirring is initiated (500 rpm), and N-Boc-hydroxylamine (1, 25.0 g, 0.188 mol, 1.3 equiv) (Note 3) is added. After stirring at room temperature (Note 4) for 5 min, isoprene (14.1 mL, 0.141 mol, 1.0 equiv) (Note 5) is added by a 24 mL plastic syringe to the round-bottomed flask over the course of 30 sec. Next, the flask is submerged to just above the solvent line in a 0 °C ice/water bath (Note 6) and stirring is continued for 10 min prior to addition of (diacetoxyiodo)benzene (60.6 g, 0.188 mol, 1.3 equiv), (Note 7) which is introduced over the course of 15 sec. The flask is capped with a 29/32 rubber septum. To prevent over-pressurization, a 5.5-cm, 18-gauge disposable needle is then inserted through the septum (Figure 1A). The reaction is stirred at 500 rpm for 30 min at 0 °C (Note 8). After the septum is removed, saturated aqueous NaHCO3 (150 mL) (Note 9) is added to the reaction solution at 0 °C over 30 sec. After the ice bath is removed, the reaction mixture is stirred at 500 rpm for 10 min at room temperature (Figure 1B). After the stirring is stopped, the stir bar is removed, and the resulting mixture is transferred to a 2 L separatory funnel. The reaction vessel is rinsed with methylene chloride (2 × 25 mL), and the resulting methylene chloride solution is transferred to the 2 L separatory funnel. Additional saturated aqueous NaHCO3 (150 mL) is added to the separatory funnel, which is then capped, shaken, and vented until the evolution of gas stops. The two layers are separated, and the aqueous layer is extracted with methylene chloride (3 × 75 mL). The organic layers are combined and washed with saturated brine (200 mL) (Note 10). The separatory funnel is shaken until the evolution of gas stops and the layers are separated. The organic layer is transferred into a 2 L Erlenmeyer flask and dried with Na2SO4 (100 g) (Note 11). After drying for 15 min, the mixture is filtered into a 2 L round-bottomed flask using a 350 mL plastic funnel plugged with cotton, and the filtrate is concentrated using a rotary evaporator (Note 12) (450 mmHg to 20 mmHg, 80 rpm, water bath temperature 38 °C). The crude product is orange/yellow oil weighing 70.4 g (Figure 1C).

Figure 1. Nitroso Diels-Alder reaction setup and purification A) Reaction cooled to 0 °C; B) Reaction mixture after quenching with saturated aqueous NaHCO3; C) The crude orange/yellow oil; D) Compound 2 after flash column chromatography (photos provided by checkers)
The crude oil is purified by flash column chromatography on silica gel using hexanes and ethyl acetate as eluents (Notes 13 and 14). This produced compound 2 (11.5 g, 41% yield, 97.2% purity, a 1.1 : 1 regioisomeric mixture) (Notes 15 and 16) as a colorless oil (Figure 1D).
B. tert-Butyl 3-methyl-1H-pyrrole-1-carboxylate (3). A three-necked, 1 L round-bottom flask (main neck: 29/32 joint, side necks: 15/25 joint) (Flask A) is equipped with a 4-cm by 1.5-cm Teflon-coated football-shaped stir bar. Two additional three-necked, 500 mL round-bottomed flasks (main neck: 29/32 joint, side necks: 15/25 joint) (Flasks B and C) are equipped with 3-cm by 1.5-cm Teflon-coated football-shaped stir bars. One side neck of each of Flasks A, B, and C is topped with a 15/25 three-way stop cock connected to an argon/vacuum line. The main neck and the other side neck of each of Flasks A, B, and C are then fitted with 29/32 and 15/25 rubber septa, respectively. All joints are sealed with grease and Teflon tape. Flasks A, B, and C are dried under vacuum (1.5 mmHg) by heating with a heat gun for 5 min, allowed to cool to room temperature, and backfilled with argon. After the 29/32 septum is removed from the main neck of Flask A, potassium tert-butoxide (11.3 g, 0.101 mol, 2.0 equiv) (Note 17) is added to Flask A through the main neck. After the 29/32 septum is removed from the main neck of Flask C, compound 2 (10.0 g, 50.2 mmol, 1.0 equiv) is added to Flask C through the main neck. Then, the removed 29/32 rubber septa are reattached to the main necks of Flasks A and C and sealed with Teflon tape. Flasks A, B, and C are evacuated under high vacuum (1.5 mmHg) for 5 min and backfilled with argon three times. Argon/vacuum lines are removed from three-way stop cocks on Flasks A, B, and C, each of which is then connected to a pair of argon inlet and outlet, respectively (Figure 2). Throughout the reaction before the addition of 2 M aqueous HCl, the argon atmosphere is maintained under continuous argon flow.

Figure 2. Anion fragmentation reaction setup of Flask A under continuous argon flow (photo provided by checkers)
Flask B is charged with diethyl ether (Note 18) in two portions (60 mL + 50 mL) by a 60 mL plastic syringe through the 29/32 septum. Diisopropylamine (15.0 mL, 0.107 mol, 2.1 equiv) (Note 19) is then added to Flask B by a 24 mL plastic syringe through the same septum. The solution in Flask B is then cooled to -78 °C using a dry ice/methanol bath (Note 20). Stirring is initiated and maintained at 500 rpm for 30 min after which time n-butyllithium (37.2 mL, 100 mmol, 2.0 equiv) (Note 21) is added dropwise over 5 min by a 60 mL plastic syringe through the 29/32 rubber septum. After the reaction mixture is stirred at 500 rpm for 30 min at -78 °C, the bath is changed to a 0 °C ice/water bath (Note 6) (Figure 3A). After the reaction mixture is stirred at 500 rpm for 30 min at 0 °C, the bath is changed to the -78 °C dry ice/methanol bath. The reaction mixture is stirred at 500 rpm for 30 min at -78 °C. Flask A containing potassium tert-butoxide is charged with diethyl ether in two portions (60 mL + 50 mL) by a 60 mL plastic syringe through the 29/32 septum. The stirred solution in Flask A (500 rpm) is cooled to -78 °C using a dry ice/methanol bath for 30 min (Note 22). Then, the reaction mixture in Flask B is slowly added to Flask A at -78 °C via a 50-cm, 10-gauge cannula through the 15/25 rubber septa (ca. 25 mL/min) (Figure 3B). After the cannula is removed, the reaction vessel (Flask A) is kept at -78 °C and stirred at 500 rpm for 1 h (Figure 3C).
During this hour, Flask C containing compound 2 is charged with diethyl ether in two portions (60 mL + 50 mL) by a 60 mL plastic syringe through the 29/32 septum. The stirred solution in Flask C (500 rpm) is cooled to -78 °C using a dry ice/methanol bath for 30 min (Note 20). The reaction mixture in Flask C is then slowly transferred (ca. 22 mL/min) into the solution of LDA/t-BuOK (LIDAKOR) in Flask A at -78 °C via a 50-cm, 10-gauge cannula through the 15/25 rubber septa. (During the transfer, the color of the solution in Flask A gradually changes from pale yellow to orange (Figure 3D). The cannula is removed, and then the reaction mixture in Flask A is stirred at 500 rpm for 30 min at -78 °C, after which the color turns dark red (Note 23) (Figure 3E). Flask A is then removed from the -78 °C bath and allowed to warm for 30 min at room temperature.

Figure 3. A) Preparation of LDA at 0 °C; B) Cannulating a solution of LDA into t-BuOK solution; C) Appearance of the LDA/t-BuOK (LIDAKOR) solution; D) Cannulating a solution of compound 2 into LIDAKOR; E) Dark red color of compound 2 treated with LIDAKOR; F) Addition of 2M aqueous HCl into the reaction mixture G) The vibrant yellow biphasic mixture; H) The crude dark brawn oil (photos provided by checkers)
After this time, the three-way stop cock and the rubber septa are removed. Then, 2 M aqueous HCl (200 mL) (Note 24) is dispensed from a 500 mL graduated cylinder and added to the reaction mixture through a 100 mL plastic funnel topped on the main neck at room temperature (Figure 3F). After the removal of the plastic funnel from the main neck, the resultant biphasic mixture is stirred at 500 rpm for 30 min, during which the color changes from red to orange and finally to a vibrant yellow (Note 25) (Figure 3G). The stir bar is removed, and the resulting reaction mixture is transferred to a 1 L separatory funnel. After the biphasic mixture is separated, the acidic aqueous layer is collected in a 1 L, 45/50 Erlenmeyer flask, and the organic layer is then washed again with 2 M aqueous HCl (200 mL) by shaking vigorously until venting results in no further gas evolution. The layers are separated, and the aqueous layers are combined. After the organic layer is collected in a separate 1 L, 45/50 Erlenmeyer flask, the combined aqueous layers are returned to the separatory funnel and extracted with diethyl ether (3 × 50 mL). All organic layers are combined, returned to the separatory funnel, and washed with saturated brine (200 mL) (Note 10) before drying with Na2SO4 (100 g) (Note 11). After 10 min at room temperature, the drying agent is removed using a 350 mL plastic funnel plugged with cotton, and the filtrate is transferred to a 2 L, 29/32 round-bottomed flask. The flask is placed on the rotary evaporator (Note 12) and the solvent is removed (580 mmHg to 20 mmHg, 80 rpm, water bath temperature 38 °C) to give 3 as a dark brown oil (10.5 g crude) (Figure 3H).
The crude product 3 is purified by flash column chromatography on silica gel using hexanes and ethyl acetate as eluents (Notes 13 and 26). This provided compound 3 (2.87 g, 32% yield, 98.9% purity) (Notes 27 and 28) as a clear-yellow oil (Figure 4).
Figure 4. Compound 3 after flash column chromatography (photo provided by checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
N-Boc-hydroxylamine,
isoprene,
(diacetoxyiodo)benzene,
dichloromethane,
diisopropyl amine,
n-butyl lithium,
potassium tert-butoxide,
diethyl ether,
methanol, dry ice,
hydrochloric acid,
sodium bicarbonate,
sodium sulfate,
hexanes,
ethyl acetate, silica gel, as well as the proper procedures for running reactions at cryogenic temperatures
.2.
Methylene chloride (>99.9%) used as solvent was obtained from Fisher Chemical and used as received (submitters).
Methylene chloride (>99.5%) was obtained from FUJIFILM Wako Pure Chemical Corporation and purified by Glass Contour solvent dispensing system (Nikko Hansen & Co., Ltd.) (checkers).
3.
N-Boc-hydroxylamine (99.8%) was obtained from Chem-Impex Int'L Inc. and used as received (submitters).
N-Boc-hydroxylamine (>97.0%) was obtained from Tokyo Chemical Industry Co., Ltd. and used as received (checkers).
4. The term "room temperature" used throughout this manuscript refers to a temperature between 22 °C to 25 °C.
5.
Isoprene (99%, stabilized with
p-tert-butylcatechol) was obtained from Sigma-Aldrich and used as received (submitters and checkers).
6. In a 2.5 L vacuum jacketed Dewar, approximately 400 g of ice and 600 mL of tap water were used to set the bath temperature at 0 °C. All temperatures reported in the procedure are bath temperatures.
7.
(Diacetoxyiodo)benzene (98%) was obtained from Oakwood Chemical and used as received (submitters).
(Diacetoxyiodo)benzene (>98.0 %) was obtained from FUJIFILM Wako Pure Chemical Corporation and used as received (checkers).
8. TLC analysis of the reaction mixture is shown below (Figure 5). Four spots on the silica gel plate (TLC Silica gel 60 F
254, obtained from Merck KGaA) can be visualized by
I2/
SiO2 stain. The TLC plate needs to be fully dipped in a dark orange mixture of
I2 (100 mg, obtained from Nacalai Tesque, Inc., used after ground by a mortar and pestle) and
SiO2 (10 g) (
Note 13) for 5 min for the purpose of visualizing the spots. The desired product
2 appears as an orange spot, the 2nd from the top, and the R
f value of
2 in
hexanes/
ethyl acetate (5/1, v/v) is 0.40.
Figure 5. TLC analysis of the reaction mixture (TM = Target Material, R = Reaction mixture) (photo provided by checkers)
9.
Sodium bicarbonate (reagent grade) was obtained from Ward's Science and used as received (submitters).
Sodium bicarbonate (>99.5%) was obtained from Nacalai Tesque, Inc. and used as received (checkers).
10.
Sodium chloride (>99.5%) was purchased from Nacalai Tesque, Inc. and used as received (checkers).
11.
Sodium sulfate (>99%) was obtained from Fisher Chemical and used as received (submitters). Anhydrous
sodium sulfate (>99.0%) was obtained from Nacalai Tesque, Inc. and used as received (checkers).
12. The rotary evaporator is a Heidolph Hei-VAP Value (submitters). The rotary evaporator was obtained from Tokyo Rikakikai Co., Ltd. (checkers).
13. The column was obtained from O'Brien's Scientific Glassware. The sand (sea washed) was obtained from Fisher Chemical and used as received. The silica gel (0.040-0.063 mm, 230-400 mesh ASTM) was obtained from Millipore and was used as received.
Ethyl acetate (>98.5%) and
hexanes (>98.5%) were obtained from Fisher Chemical and used as received. (submitters).
The column was obtained from Sansyo Co., Ltd. The sea sand was obtained from Nacalai Tesque, Inc. and used as received. The silica gel (Silica gel 60 N, 0.040-0.050 mm, spherical and neutral) was obtained from Kanto Chemical Co., Inc. and was used as received. Ethyl acetate (>99.0%) and hexanes (>96.0%) were obtained from Kanto Chemical Co., Inc. and used as received (checkers).
14. Flash column chromatography is performed under compressed air. A 14-cm by 50-cm flash chromatography column with no solvent bulb is fitted with a cotton plug. A 2 cm bed of sand is added and the column is wet packed with 700 g of silica gel in
hexanes (1.0 L). Sea sand with 3 cm minimum height is added to the top of the column, then the crude product is loaded onto the column as a neat oil. Fraction collection is then started using 500 mL glass bottles to collect the eluent. Elution is continued with 1500 mL of
hexanes/
ethyl acetate (1/0, v/v), 1500 mL of
hexanes/
ethyl acetate (30/1, v/v), 4500 mL of
hexanes/
ethyl acetate (20/1, v/v), and then 2500 mL of
hexanes/
ethyl acetate (10/1, v/v). As illustrated in Figure 6, the fractions containing the product (
2) are identified as fractions No. 10 through No. 18 by TLC (R
f of 0.40,
hexanes/
ethyl acetate = 5/1 (v/v), visualized by
I2/
SiO2 stain). These fractions are combined, concentrated using a rotary evaporator (
Note 12) (180 mmHg to 20 mmHg, 80 rpm, water bath temperature 38 °C), and then placed under high vacuum (1.5 mmHg, room temperature) for 1 h to remove residual solvent.

Figure 6. TLC analysis of column chromatography (photo provided by checkers)
15. The identity of a 1.1 : 1 regioisomeric mixture of compound
2 was established with the following characterization data.
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.497 (s, 0.52 × 9H), 1.503 (s, 0.48 × 9H), 1.66 (s, 0.52 × 3H), 1.73 (s, 0.48 × 3H), 3.94 (s, 0.48 × 2H), 4.03 (s, 0.52 × 2H), 4.26 (s, 0.52 × 2H), 4.36 (s, 0.48 × 2H), 5.52 (m, 1H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 18.5 (0.48 × 1C), 19.9 (0.52 × 1C), 28.4 (0.52 × 3C), 28.5 (0.48 × 3C), 44.9 (0.52 × C), 48.7 (0.48 × C), 68.2 (0.48 × C), 71.3 (0.52 × C), 81.7 (1C), 116.7 (0.52 × C), 118.2 (0.48 × C), 130.5 (0.48 × C), 131.7 (0.52 × C), 155.1 (0.48 × C), 155.2 (0.52 × C); HRMS (ESI+) calculated for C
10H
17NNaO
3 [M+Na]
+ 222.1101, found 222.1101; IR (KBr film): 2977 (w), 2932 (w), 2853 (w), 1727 (m), 1704 (s), 1476 (w), 1443 (w), 1390 (m), 1367 (s), 1240 (m), 1165 (s), 1098 (m), 1059 (w), 1040 (w), 1013 (w), 958 (w), 904 (w), 857 (w), 832 (w), 762 (w), 716 (w), 555 (w), 517 (w), 443 (w) cm
-1; TLC: R
f = 0.40 in
hexanes/
ethyl acetate (5/1, v/v). The purity of compound
2 was calculated to be >97% by qNMR
pdf with the relaxation delay set to 30 seconds using 11.0 mg of
1,3,5-trimethoxybenzene (FUJIFILM Wako Pure Chemical Corporation, >99.0%, used as received) and 11.8 mg of compound
2.
16. When the reaction was carried out on a 70.5 mmol scale, 6.09 g (43%) of compound
2 was obtained with 98.8% purity.
17.
Potassium tert-butoxide (≥98%) was obtained from Sigma-Aldrich and used as received (submitters and checkers). Two equivalents of the LIDAKOR base are used due to incomplete conversion of starting material when only one equivalent is used.
18.
Diethyl ether (>99%) was obtained from Fisher Chemical and was dried using a solvent purification system manufactured by SG Water U.S.A., LLC (submitters).
Diethyl ether (>99.5%) was obtained from FUJIFILM Wako Pure Chemical Corporation and purified by Glass Contour solvent dispensing system (Nikko Hansen & Co., Ltd.) (checkers).
19.
Diisopropylamine (99%) was obtained from Oakwood Chemical and was dried over
calcium hydride and freshly distilled (submitters).
Diisopropylamine (>99.0%) was obtained from Tokyo Chemical Industry Co., Ltd. and was dried over
calcium hydride and freshly distilled (checkers).
20. In a 1 L vacuum jacketed Dewar, approximately 500 g of dry ice and 250 mL of
methanol were used to set the bath temperature at -78 °C.
21.
n-Butyllithium (2.5 M in
hexanes) was obtained from Sigma-Aldrich and used as received (submitters).
n-Butyllithium (2.69 M in
n-hexanes) was obtained from Kanto Chemical Co., Inc. and used as received (checkers).
22. In a 2 L vacuum jacketed Dewar, approximately 750 g of dry ice and 400 mL of
methanol were used to set the bath temperature at -78 °C.
23. TLC analysis of the reaction mixture using
hexanes/
ethyl acetate (5/1, v/v) as eluent is shown below (Figure 7). The progress of the reaction can be followed by observing the loss of starting material
2, and the formation of intermediates
8 (see Discussion section, Scheme 3). These spots on the silica gel plate (TLC Silica gel 60 F
254, obtained from Merck KGaA) can be visualized by
I2/
SiO2 stain.
Figure 7. TLC analysis of the reaction mixture (SM = Start Material, R = Reaction mixture) (photo provided by Checkers)
24.
Hydrochloric acid (2.5 L, 37% (12 M) aqueous solution) was obtained from Fisher Chemical and diluted to a concentration of 2 M using DI water (submitters).
Hydrochloric acid (4.0 L, 37% (12 M) aqueous solution) was obtained from FUJIFILM Wako Pure Chemical Corporation and diluted to a concentration of 2 M using pure water (FUJIFILM Wako Pure Chemical Corporation) (checkers).
25. TLC analysis of the reaction mixture is shown below (Figure 8). The R
f value of the desired product
3 in
hexanes/
ethyl acetate (5/1, v/v) is 0.60. The spot of compound
3 on the silica gel plate (TLC Silica gel 60 F
254, obtained from Merck KGaA) can be visualized by UV light (254 nm) and
I2/
SiO2 stain.
Figure 8. TLC analysis of the reaction mixture (TM = Target Material, R = Reaction mixture) (photos provided by checkers)
26. Flash column chromatography is performed under compressed air. A 8-cm by 32-cm flash chromatography column with no solvent bulb is fitted with a cotton plug and a 3 cm bed of sand is added. The column is then wet packed with 500 g of silica gel in
hexanes (800 mL) and sea sand (3 cm height) is added to the top of the column. Then, the crude product is loaded onto the column as a neat oil. Fraction collection is then started using 500 mL glass bottles. Elution is continued with 2500 mL of
hexanes/
ethyl acetate (1/0, v/v), 2500 mL of
hexanes/
ethyl acetate (50/1, v/v), 2500 mL of
hexanes/
ethyl acetate (35/1, v/v), and then 5000 mL of
hexanes/
ethyl acetate (25/1, v/v). The location of product
3 as determined by TLC (R
f of 0.60,
hexanes/
ethyl acetate = 5/1 (v/v), visualized by UV light (254 nm)) is determined to be fractions No.12 through No. 16 (Figure 9). These fractions are combined, concentrated using a rotary evaporator (
Note 12) (180 mmHg to 20 mmHg, 80 rpm, water bath temperature 38 °C), and dried on a high vacuum (1.5 mmHg, room temperature) for 1 h.

Figure 9: TLC analysis of column chromatography (photo provided by checkers)
27. The identity of compound
3 was established with the following characterization data.
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.58 (s, 9H), 2.06 (s, 3H), 6.05 (s, 1H), 6.97 (s, 1H), 7.14 (s, 1H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 12.0, 28.1 (3C), 83.2, 114.1, 117.3, 120.1, 122.1, 149.1; HRMS (ESI+) calculated for C
10H
15NNaO
2 [M+Na]
+ 204.0995, found 204.0996; IR (KBr film): 2979 (w), 2930 (w), 1741 (s), 1487 (w), 1456 (w), 1398 (s), 1368 (m), 1344 (s), 1251 (s), 1164 (s), 1120 (m), 1069 (m), 1035 (w), 997 (w), 971 (s), 853 (w), 826 (w), 773 (s), 708 (w), 616 (w), 586 (w), 546 (w), 526 (w), 481 (w), 462 (w), 411 (w) cm
-1; TLC: R
f = 0.60 in
hexanes/
ethyl acetate (5/1, v/v). The purity of compound
3 was calculated to be >98% by qNMR
pdf with the relaxation delay set to 30 seconds using 16.5 mg of
1,3,5-trimethoxybenzene (FUJIFILM Wako Pure Chemical Corporation, ≥99.0%, used as received) and 16.0 mg of compound
3.
28. When the reaction was carried out on a 28.1 mmol scale, 1.73 g (34%) of compound
3 was obtained with 97.9% purity.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The pyrrole moiety is found in a wide range of chemical compounds including, but not limited to, natural products, therapeutics, polymers, and dyes.
2 Moreover, these privileged heterocycles can participate in a number of useful carbon-carbon bond-forming reactions, thereby rendering them valuable synthetic intermediates.
3 In our recent synthetic study directed toward longeracemine (
4),
4 we identified
N-Boc-3-methylpyrrole (
3) as a potential starting material. As illustrated in Scheme 1, we envisioned constructing the 2-azabicyclo[2.2.1] heptane framework of
4 through a SmI
2-mediated spirocyclization/rearrangement cascade initiated from an intermediate Diels-Alder adduct (
6). Although commercially available, the cost of 3-methylpyrrole ($800/g from Millipore Sigma) was prohibitive, particularly given its planned use in the early stages of what was certain to be a challenging synthetic endeavor.
Scheme 1. Retrosynthetic analysis of longeracemine and motivation for present study
Turning instead to literature preparations, we noted that previous syntheses of
3 have taken many forms. For example, Castagnolo and coworkers reported a one-step tandem enyne cross metathesis/cyclization to access various protected 3-methylpyrroles.
5 Although efficient, the relatively high loading (5-10 mol%) of the Grubbs 2
nd Generation catalyst rendered this approach cost-prohibitive.
6 Lash also reported a robust synthesis of 2-carboxy-4-methylpyrrole-starting from glycine ethyl ester, however the sequence is 4-steps followed by decarboxylation.
7 Lancaster and VanderWerf have also reported a 4-step sequence involving condensation of aminoacetone with diethyl oxalacetate and subsequent hydrolysis and decarboxylation of the product, 2-carboxy-3-carbethoxy-4-methylpyrrole.
8 After dismissing the above syntheses on the basis of cost, step-count, or practicality we came across an intriguing nitroso Diels-Alder approach reported by Kouklovsky.
9 As described in Scheme 2, Kouklovsky's sequence begins with
in situ formation of the acyl nitroso dienophile derived from
1 via oxidation (
e.g., NaIO
4) in the presence of an appropriate diene such as isoprene. This latter reaction results in the formation of a regioisomeric mixture of dihydrooxazines (
2), via [4+2] cycloaddition, which is then subjected to conditions suited for reductive N-O bond cleavage. Derived alcohols
7 are oxidized to the corresponding carbonyls and advanced via cyclo-condensation and aromatization to furnish
3. Although we found the relative step- and cost-efficiency of this latter approach attractive, we thought it worthwhile to spend some time optimizing the sequence. To this end, we first found phenyliodine diacetate (PIDA), in lieu of NaIO
4 or Bu
4NIO
4, to be a preferred oxidant. We next turned to exploring alternative methods for reductive N-O bond cleavage, so as to avoid using super-stoichiometric quantities of highly toxic Mo(CO)
6. In initial investigations, we established that single electron transfer agents, such as SmI
2,
10 were effective but gave inconsistent results and poor yields of
7. Further investigation into other methods eventually led to our exploration N-O bond cleavage by simple treatment with strong base followed by addition of a reductant such as lithium aluminum hydride (LAH). As shown in Scheme 3, we reasoned that deprotonation at either allylic position could result in N-O bond cleavage to form a corresponding anionic intermediate (
8) which, upon subsequent reduction could give
7. Interestingly, when exploring solvents for the reaction, we noted trace quantities of the desired product (
3) were observed when Et
2O was used instead of THF, presumably via ring-closure of the amide anion, prior to the introduction of LAH, and eventual aromatization upon work-up. Given that this latter transformation preserved the oxidation level of the cycloadduct, thereby obviating the subsequent reduction/oxidation sequence, we performed an extensive optimization study. To this end, screening of over 20 different bases eventually revealed that exposing the mixture of dihydrooxazines (
2) to a solution of LDA/
t-BuOK (LIDAKOR
11) in Et
2O at -78 °C resulted in reasonable and consistent yields of
3.
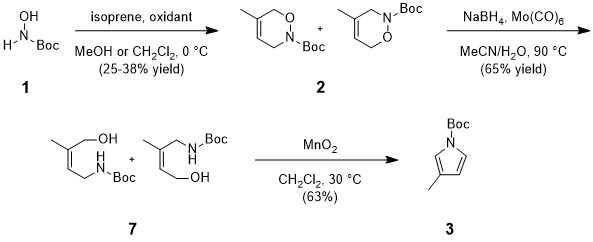
Scheme 2. Outline of Kouklovsky's reported synthesis of 3
We have detailed the development of a two-step synthesis of N-Boc-3-methylpyrrole (3). The synthesis proceeds through a Diels-Alder reaction between isoprene and the nitroso dienophile derived from 1 via oxidation with PIDA. Treatment of the resulting regioisomeric mixture of dihydrooxazines 2 with LIDAKOR in Et2O at -78 °C furnishes 3. The synthesis employs inexpensive and non-toxic reagents and is anticipated to expand the use of this privileged heterocycle through broadened access.
Scheme 3. Advancing 2 by treatment with base
Appendix
Chemical Abstracts Nomenclature (Registry Number)
N-Boc-hydroxylamine: tert-Butyl N-hydroxycarbamate; (1) (36016-38-3)
Isoprene: (78-79-5)
(Diacetoxyiodo)benzene: Iodobenzene diacetate (3240-34-4)
Methylene chloride: Dichloromethane; (75-09-2)
Diethyl ether: (60-29-7)
Diisopropylamine: (108-18-9)
n-Butyllithium: Butyllithium; (109-72-8)
Potassium tert-butoxide: (865-47-4)

|
Daniel P. Graeff was born in San Diego, CA. He received his B.S. in Chemistry with a minor in Business Administration from Baylor University in 2022. While at Baylor he worked on reaction development and total synthesis under the mentorship of Dr. John L. Wood. He is now working towards his Ph.D. in Chemistry at Rice University. |

|
Dr. Noah J. Sims earned his B.S. in Chemistry from the University of Rochester in 2018 under the tutelage of Dr. Robert K. Boeckman, Jr. Soon after, he moved to Waco, TX where he continued to pursue his love for total synthesis under the mentorship of Dr. John L. Wood. His research is focused on efficient construction of complex natural products and resulted in completed syntheses of caesalpinnone A, caesalpinflavan B, and alterbrassicicene C. Noah moved on to postdoctoral studies at Caltech. |

|
Quentin R. Savage received his B.S. in Chemistry and minor in Physics from the University of Tennessee at Martin in 2019 where he performed academic research under supervision of Dr. Phillip Shelton studying green organic chemistry methods development. Quentin is currently pursuing his Ph.D. in the laboratory of Dr. John Wood at Baylor University. His research is primarily focused on the total synthesis of complex natural products. |

|
Dr. Richard K. Jackson, III received his B.S. in Chemistry from the University of Dallas in 2014. He completed his Ph.D. at Baylor University where, under the mentorship of John L. Wood, his research focused on the total synthesis of complex natural products and completed the synthesis of ent-plagiochianin B. Following receipt of the Ph.D., Ricky took a position at FMC corporation. |

|
Dr. Amy C. Jackson received her B.S. degree in Chemistry in 2018 from San Diego State University and her Ph.D. from Baylor University in 2022 where, under the supervision of Dr. John L. Wood, she focused on developing new strategies for complex molecule synthesis and completed the total synthesis of (+)-raistrickindole A. Following her receipt of the Ph.D., Amy took the position of Science Advisor at Goodwin Law. |

|
Danielle M. Lawson is currently pursuing her B.S. in Biology with a minor in Biochemistry from Baylor University where she plans on graduating in the spring of 2023. Dani continues to work on total synthesis under the mentorship of Dr. John L. Wood. After graduation, Dani plans to continue her education and pursue her Ph.D. in Biochemistry. |

|
Madelyn R. Wixom is currently working toward a B.S. in Forensic Science at Maryville University, St. Louis. She has performed undergraduate research focusing on the total synthesis of complex natural products in the laboratory of Dr. John L. Wood at Baylor University. |

|
John L. Wood received a B.A. degree from the University of Colorado in 1985 and a Ph.D. from the University of Pennsylvania in 1991 under the direction of Amos B. Smith, III. In 1991 he moved to Harvard University as an American Cancer Society postdoctoral fellow and continued studying natural products synthesis in the laboratories of Stuart Schreiber. He joined the faculty at Yale University in 1993 as an Assistant Professor and was promoted to Full Professor in 1998. In 2006, Professor Wood joined the faculty at Colorado State University as the Albert I. Meyers Professor of Chemistry and in 2013 moved to Baylor University where he serves as the Robert A. Welch Distinguished Professor of Chemistry and Department Chair. The focus of Professor Wood's research is synthetic organic chemistry with an emphasis on designing innovative solutions to problems in natural product synthesis. |

|
Yuya Shiga was born in Saitama, Japan. He graduated from the University of Tokyo in 2022 with B.S. in Pharmaceutical Science. He is continuing his graduate studies at the University of Tokyo under the supervision of Prof. Masayuki Inoue. His research interests are in the area of the total synthesis of complex natural products. |

|
Haruka Fujino received his Ph.D. (2019) from The University of Tokyo under the supervision of Prof. Masayuki Inoue. During the Ph.D. course, he spent 2 months as Visiting Student at the University of Chicago under the direction of Prof. Scott A. Snyder (2016). After carrying out postdoctoral research with Prof. Seth B. Herzon at Yale University (2019-2020), he was appointed as Assistant Professor in the Graduate School of Pharmaceutical Sciences at the University of Tokyo in 2020. His research interests include the total synthesis of bioactive and architecturally-complex natural products. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved