Org. Synth. 2023, 100, 218-233
DOI: 10.15227/orgsyn.100.0218
Synthesis of Potassium 5-Bromopentyltrifluoroborate via Hydroboration of a Haloalkene
Submitted by Sarah J. Burke, Latifah M. Alhthlol, James M. Gamrat, Giulia Mancini, Christopher L. Orme, Jacqueline R. Santhouse, Dylan T. Tomares, and John W. Tomsho*
1Checked by Philip Boehm and Sarah E. Reisman
1. Procedure (Note 1)
Potassium 5-bromopentyltrifluoroborate (3). An oven-dried three-necked, 250 mL round-bottomed flask is fitted with a gas inlet and a thermocouple inserted through a rubber septum. The flask is equipped with a 2.25 cm Teflon-coated magnetic stir bar, and the third neck fitted with a rubber septum. The flask is evacuated (<0.1 mmHg) and backfilled with nitrogen three times. The flask is charged with 5-bromo-1-pentene (4.8 mL, 40.4 mmol, 1.0 equiv) (Note 2) and triethylsilane (6.9 mL, 42.9 mmol, 1.06 equiv) (Note 3), both via syringe. While stirring, the mixture is cooled to -78 ℃ with a dry ice-acetone bath (Figure 1A), (Note 4) followed by the dropwise (3 mL/min) addition of a 1.0 M solution of boron trichloride in hexane (46 mL, 46 mmol, 1.1 equiv) (Notes 5 and 6) via glass syringe. The reaction is stirred at -78 ℃ for 40 min, the dry ice-acetone bath is removed, and the reaction is stirred for an additional 2 h (Figure 1B). Prior to the aqueous quench, the reaction mixture is cooled to 0 ℃ with an ice bath. The rubber septum is removed, and water (50 mL) is added in two equal portions over the course of five min to convert the dichloroborane to the corresponding boronic acid (Note 7). The ice bath is removed, and after stirring for 30 min the reaction mixture is transferred to a 250 mL separatory funnel and extracted with ether (3 x 100 mL) (Note 4). The combined organics are washed with brine (100 mL) in a 500 mL separatory funnel, dried over anhydrous sodium sulfate (60 g), and filtered using a Büchner funnel (9.0 cm) with ether rinsing (20 mL) (Note 4). The organic solvent is transferred to a 500 mL round-bottomed flask and concentrated on the rotary evaporator (~1-2 mmHg) followed by a high vacuum pump (0.01 mmHg). A clear, colorless oily residue (disiloxane) with a white solid (boronic acid) is formed (Figure 2) (Note 8).

Figure 1. (a) Reaction mixture after adding all reactants and (b) reaction mixture after removing the dry-ice acetone bath (photos provided by checker)
Figure 2. Boronic acid (white solid) in disiloxane by-product (clear, colorless oil) (photo provided by submitter)
To the mixture of boronic acid and disiloxane byproduct, diethyl ether (100 mL) along with a 2.25 cm Teflon coated stir-bar is added, followed by potassium hydrogen difluoride (13.3 g, 169.8 mmol, 4.2 equiv) (Note 9). The flask's contents are stirred, and water (2 mL) is added via syringe in ~0.5 mL portions every 15 min (Figure 3). This mixture is stirred for an additional 1 h at 23-25 ℃ (Note 10), after which time the mixture is filtered through a Büchner funnel (9.0 cm) while rinsing the flask and the funnel with acetone (1 x 100 mL) (Note 4) to remove any byproduct salts. The filtrate is transferred to a 250 mL round-bottomed flask and concentrated on the rotary evaporator (~1-2 mmHg). The resulting solid is purified by dissolution in a minimum of acetone (ca. 10 mL) and precipitation via slow addition of this solution of the trifluoroborate salt into diethyl ether (500 mL) (Note 4). Precipitation occurs immediately. The precipitate is collected using a Büchner funnel (9.0 cm), and the solid is rinsed with diethyl ether (3 x 20 mL) (Note 4). The solid is dried for at least 1 h under high vacuum (0.01 mmHg) to provide 7.79 g (75%) of pure 3 (Notes 11, 12, and 13) (Figure 4).

Figure 3. Conversion of the boronic acid to the corresponding trifluoroborate salt (photo provided by submitter)
Figure 4. Final product, crystalline trifluoroborate salt (photo provided by submitter)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as in Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011); the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-thelaboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of the current procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
5-bromo-1-pentene,
triethylsilane,
boron trichloride,
potassium hydrogen difluoride,
hexane,
diethyl ether,
acetone, dry ice,
DMSO-d6, and
acetone-d6.
2.
5-Bromo-1-pentene (98%) was obtained from Oakwood Chemical (013455) and used as received.
3.
Triethylsilane (99%, AcroSeal, ACROS Organics) was obtained from Fisher Scientific and used without further purification.
4. All solvents were purchased from major suppliers and used without further purification. Specifically:
acetone, HPLC-grade, Sigma-Aldrich;
diethyl ether, reagent grade, VWR;
acetone-d6, >99.8% D, Sigma-Aldrich.
5.
Boron trichloride, 1M solution in
hexane, (AcroSeal, ACROS Organic) was obtained from Fisher Scientific and used as received. This reagent fumes in air as it forms
hydrogen chloride on contact with moisture. We recommend working quickly with
boron trichloride, and washing all syringes immediately because the residue inside tends to become very sticky. Comparable yields are observed when employing 1M
boron trichloride in solvents other than
hexane, e.g.,
dichloromethane and
heptane, in otherwise identical procedures.
6. A temperature increase of approximately 1 ℃ per minute is observed upon addition of
boron trichloride. This temperature increase does not negatively impact the reaction outcome.
7. The addition of water to convert the dichloroborane to the boronic acid product is very exothermic. It also causes foaming of the reaction mixture and formation of significant
hydrogen chloride gas as evidenced by smoking. This step should be performed slowly and carefully in a fume hood.
8. The chemical formula for the
disiloxane byproduct is
(Et3Si)2O. This clear, colorless, oily substance has a high boiling point and very low reactivity. It is carried through the next step where it is subsequently removed via filtration of the product.
9.
Potassium hydrogen difluoride (99%) was obtained from Sigma-Aldrich and used as received. This reagent will permanently etch glassware.
10. Reactions are found to have gone to completion after 2 h reaction time, as determined by
1H NMR. No loss of yield is observed if reaction is left to proceed for up to 24 h.
11. White shimmery solid. mp > 250 ℃.
1H NMR
pdf (400 MHz,
acetone-d6) δ: 3.45 (t,
J = 7.0 Hz, 2H), 2.03-1.63 (m, 2H), 1.32 (dq,
J = 32.3, 8.3, 7.9 Hz, 4H), 0.18 (h,
J = 7.0 Hz, 2H).
13C NMR
pdf (101 MHz,
acetone-d6) δ: 35.5, 34.1, 32.6, 25.1 (q, J = 2.5 Hz), 19.0 (br).
11B NMR
pdf (128 MHz,
acetone-d6) δ: 5.67.
19F NMR
pdf (376 MHz,
acetone-d6) δ: -141.06. IR (neat): 2910 (s), 2840 (s), 1239 (s), 1078 (s), 1037 (s), 921 (s), 861 (s) cm
-1. HRMS (ESI)
m/z calcd for C
7H
13BNF
2Br [M - (KF) + (CH
3CN)]- 239.0287, found 239.0294.
12. Analysis by qNMR
pdf, performed with
1,3,5-trimethoxybenzene (Millipore Sigma, >99%) as an internal standard, indicated that the purity of the product was 99%.
13. A second run completed on the same scale gave 7.73 g product (75% yield, 98% purity by qNMR).
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Boronic acids and their derivatives have recently emerged as biologically interesting moieties due, in part, to their increased use in the pharmaceutical industry.
2 To date the FDA has approved five drugs containing boron: Velcade®, a peptidyl boronic acid treatment for multiple myeloma;
3 Kerydin
TM, an oxaborole-containing antifungal;
4 Ninlaro®, a reversible proteasome inhibitor for the treatment of multiple myeloma;
5 Eucrisa
TM, a PDE-4 inhibitor used for the treatment of atopic dermatitis;
6 and Vabomere
TM, a β-lactamase inhibitor.
2c The incorporation of boron in pharmaceutical agents has been motivated by the boron atom's Lewis acidic character. Under physiologically relevant conditions, a neutral, trigonal planar boron center contains a vacant p-orbital that may interact with Lewis bases. Boron's vacant p-orbital has been shown to engage in reversible, dative bonding with Lewis basic amino acids in the active sites of enzymes to form anionic, tetrahedral boron-enzyme complexes.
2While investigating nucleophilic substitution reactions involving boron-containing electrophiles as a method to synthesize new pharmaceutical agents, we found boron's vacant p-orbital caused problems in our syntheses. To overcome these difficulties, trifluoroborate salt electrophiles were explored as a method to mask the vacant p-orbital. Because these reagents are not commercially available and their corresponding boronic acids are relatively expensive, we envisioned synthesizing potassium haloalkyltrifluoroborate salts (e.g., potassium 5-bromopentyltrifluoroborate salt
3) via hydroboration of commercially available haloalkenes (e.g., 5-bromo-1-pentene
1). Reports of the hydroboration of haloalkenes have been related by others, but the drawbacks of these reactions include the use of large boronate esters (i.e., pinacol) which lack atom economy, low yields and/or irreproducibility, expensive reagents, inadequate experimental details, or unpurified reaction products.
7 In fact, all our initial synthetic attempts using literature procedures were problematic. Reactions with Wilkinson's catalyst and pinacolborane resulted in decomposition, likely caused by poor catalyst chemoselectivity.
8 Success was realized with [Ir(cod)Cl]
2 and pinacolborane, but high cost and modest yields discouraged further consideration.
7i Additionally, use of catecholborane as the hydroboration reagent afforded good yields of the hydroboration product.
7e However, hydrolysis or transesterification with pinacol presented catechol and benzoquinone contamination, which have been widely reported in the literature.
7 Finally, use of commercially available dichloroborane dioxane
9 and dibromoborane dimethyl sulfide
10 complexes resulted in low yields and a borinic acid byproduct. As discussed by Brown, borinic acid byproducts are caused by disproportionation of the dihaloborane complexes.
9aDichloroborane was next examined as the hydroboration reagent. To avoid disproportionation, dichloroborane was prepared in situ from boron trichloride and triethylsilane.
7b This reagent provided moderate to low yields of boronic acids (
2, Scheme 1) after aqueous work-up and purification via partitioning, recrystallization, or chromatography. Low yields were caused by a hydroboration byproduct that is rarely mentioned in the literature: 1,1,1,3,3,3-hexaethyldisiloxane, (Et
3Si)
2O. It was observed that the boronic acid products were soluble in this byproduct, making purification challenging. In only one case, boronic acid
2, was an acceptable yield (79%) of the hydroboration product obtained, versus
2b and
2c at 27% and 22% yields respectively. The five carbon alkyl chain may have resulted in decreased solubility of
2 in water compared to
2b and
2c, allowing for an efficient recrystallization. As expected, treatment of these boronic acids with potassium hydrogen difluoride as previously related by Vedejs
11 and Molander
12 (Scheme 1) proceeded without difficulty in moderate to high yields (62-90%).
13
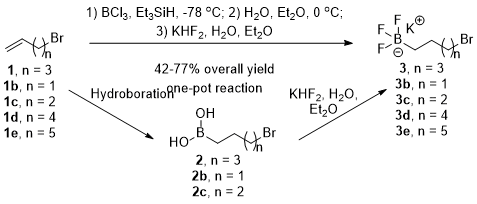
Scheme 1. The reported high yielding, sequential synthesis of potassium haloalkyltrifluoroborate salts (top) compared to a lower yielding, two-step procedure where the boronic acid intermediates are isolated (bottom)
Though successful in producing the desired products, this two-step procedure (Scheme 1, bottom) resulted in low overall yields (14-71%). In an effort to improve the yields of this series of reactions, several modifications of this general procedure were examined. The problematic disiloxane byproduct was expected to be unreactive to the reaction conditions required for the formation of the trifluoroborate salt (KHF2, H2O, ether) so the difficult purification of the intermediate boronic acid products (2) was eliminated. In this sequential, two-pot procedure, crude boronic acid products contaminated with disiloxane byproduct were isolated via an aqueous workup and carried forward for the conversion to the potassium trifluoroborate salt. Once these salts were formed, removal of the disiloxane byproduct was achieved via trituration of the crystalline products with ether.
This general sequential methodology, reported in detail herein for the conversion of 5-bromo-1-pentene (1) to the potassium 5-bromopentyltrifluoroborate salt (3), was additionally tested by the authors on a series of related haloalkene starting materials (1b-e). In all cases, this streamlined methodology provides significantly higher yields of the desired potassium haloalkyltrifluoroborate salts (3b-e, Table 1). Additionally, this is the first reported synthesis of potassium 7-bromoheptyltrifluoroborate (3e). Collectively, these results show that the sequential procedure provides a quick, reliable, and effective method for the production of potassium haloalkyltrifluoroborate salts in comparison to those found in the literature.
Table 1. Results of the application of the checked protocol on a series of related haloalkenes
a Reaction scales ranged from 34.5 - 45.2 mmol. Yields reported reflect isolated yields with purity > 96% via q-NMR. b Data from checked procedure included for comparison
In conclusion, we have developed and reported a convenient,
sequential synthesis
13 as a general method for the preparation of potassium haloalkyltrifluoroborate salts through hydroboration with dichloroborane followed by treatment of the crude hydroboration products with potassium hydrogen difluoride. The reported procedure provided high yields and easy separation from disiloxane, a rarely mentioned hydroboration byproduct. This technique improves upon those that have been reported in terms of yield, reproducibility, atom economy, convenience, and cost for the generation of a series of potassium haloalkyltrifluoroborate salts.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Triethylsilane; (617-86-7)
Boron trichloride; (10294-34-5)
Potassium hydrogen difluoride (potassium fluoride); (7789-29-9)
5-Bromo-1-pentene; (1119-51-3)
Potassium 5-bromopentyltrifluoroborate; (3) (888711-62-4)

|
Professor Sarah Burke completed her undergraduate studies at Cabrini College and earned her Ph.D. from Bryn Mawr College under the supervision of Dr. William Malachowski. After completing her post-doctoral work with Dr. John W. Tomsho at the University of the Sciences in Philadelphia, she began her academic career teaching chemistry at Neumann University. |

|
Latifah M. Alhthlol received her undergraduate degree at King Faisal University (KFU) in 2010. She then completed her M.S. at Middle Tennessee State University (MTSU) in inorganic chemistry in 2016. Currently, she is a Ph.D. in Chemistry candidate at the University of the Sciences in Philadelphia working on the synthesis of small molecules to inhibit HIV under the guidance of Dr. John W. Tomsho. |

|
Dr. James M. Gamrat obtained his B.S. degree in chemistry from Muhlenberg College in 2013. He then went on to earn his Ph.D. in chemistry in the lab of Dr. John W. Tomsho at the University of the Sciences in Philadelphia in 2021. He is currently a postdoctoral fellow in the lab of Dr. Jason Wallach at the University of the Sciences in Philadelphia. |
|
Dr. Giulia Mancini completed her undergraduate degree at University of Bologna in 2012, where she also obtained her M.S. in chemistry in 2015. In 2020, she earned her Ph.D. in Chemistry at the University of the Sciences in Philadelphia working in Dr. John W. Tomsho's group. She is currently doing postdoctoral work at the University of Basel. |

|
Christopher L. Orme obtained his M.S. in Medicinal Chemistry at The University of York (UK) in 2016. He completed his Masters project in natural product total synthesis at Victoria University of Wellington (NZ) before embarking on Ph.D. research in Chemistry at the University of the Sciences in Philadelphia (USA) under the guidance of Dr. John W. Tomsho. |

|
Jacqueline R. Santhouse is a Ph.D. candidate at the University of Pittsburgh working with peptidomimetics and unnatural peptide backbones. She received her Bachelor's in Pharmaceutical Chemistry from The University of the Sciences. |

|
Dylan T. Tomares obtained his B.S. degree in Biochemistry from the University of the Sciences in 2016. He is pursuing his Ph.D. in Chemistry at the University of Pittsburgh studying under Dr. W. Seth Childers. |

|
Professor John W. Tomsho obtained his B.S. in Pharmaceutical Chemistry at the Philadelphia College of Pharmacy and Science. His doctoral work in Medicinal Chemistry was done under the supervision of Dr. James K. Coward at the University of Michigan and his postdoctoral work under Dr. Stephen J. Benkovic at the Pennsylvania State University. He has been a Professor of Chemistry & Biochemistry at the University of the Sciences in Philadelphia since 2012. |

|
Dr. Philip Boehm did his undergraduate studies in chemistry at the Technical University Munich, Germany, and at the University of California, Berkeley, where he worked in the labs of Prof. John Hartwig. In 2022, he completed his PhD with Prof. Bill Morandi at ETH Zürich, working on the development of reversible transition metal-catalyzed reactions. He then joined the group of Prof. Sarah Reisman for a postdoc, and he is currently working on the synthesis of complex natural products. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved