1. Procedure (Note 1)
B. Acetophenone O-acetyl oxime (3). A 100 mL single-necked (15/25) round-bottomed flask equipped with a football-shaped Teflon-coated magnetic stir bar (10 × 25 mm) is charged with the crude oxime 2 (3.29 g). Acetic anhydride (3.90 mL, 41.3 mmol, 2.0 equiv) is added by a plastic syringe (Note 12) (Figure 3A). The flask is fitted with a water-cooled Dimroth condenser (20 cm, 15/25 joint) and the juncture of the glassware is sealed with Teflon tape. The reaction vessel is placed in a pre-heated silicon oil bath (100 ℃, oil bath temperature) (Notes 13 and 14) (Figure 3B). The color of the solution, which is open to the air, changes to brown after stirring for 3 h at 100 ℃. The reaction vessel is then removed from the oil bath and cooled to 23 ℃ over 30 min (Figure 3C).
Figure 3. A) Reaction mixture after the addition of acetic anhydride; B) Reaction setup; C) Reaction mixture after 3 h (photos provided by checkers)
Ethyl acetate (20 mL) and water (50 mL) are added to the flask and the resultant mixture is then transferred to a 200 mL separatory funnel, rinsing the flask with ethyl acetate (3 x 10 mL) (Figure 4A). The two layers are separated, and the aqueous layer is further extracted with ethyl acetate (2 x 40 mL). The combined organic layers are dried over anhydrous MgSO4 (10 g) for 5 min and filtered through a glass funnel with a cotton plug into 500 mL round-bottomed flask (Figure 4B). The filter cake is rinsed with ethyl acetate (2 x 20 mL), and the filtrate is concentrated with a rotary evaporator under reduced pressure (46 ℃, 12 mmHg) for 30 min. The resulting brown oil is dried under reduced pressure (23 ℃, 4.0 mmHg) for 1 h to give the crude mixture as a brown solid (Figure 4C).
Figure 4. A) The organic layer and the aqueous layer; B) Cotton filtration of MgSO4 after extraction; C) Crude brown solid (photos provided by checkers)
The crude residue is recrystallized from hexane/ethyl acetate (5/1) (Figure 5A) to afford acetophenoneO-acetyl oxime (3) (2.51 g, 99.5% purity) as a white solid (Note 15) (Figure 5B). The mother liquor is concentrated by a rotary evaporator under reduced pressure (38 ℃, 18 mmHg) for 10 min to afford the crude residue as a brown oil (Figure 5C). The crude residue is purified by flash column chromatography on silica gel using hexane and ethyl acetate as eluents (Note 16) (Figure 5D) to afford acetophenoneO-acetyl oxime (3) (791 mg, 99.5% purity) as a white solid (Note 17) (Figure 5E). In total, 3.30 g of acetophenoneO-acetyl oxime (3) is obtained (90% yield over two steps from compound 1) (Note 18).
Figure 5. A) Filtration set-up after rinsing with hexane; B) AcetophenoneO-acetyl oxime (3) obtained by recrystallization; C) Concentrated mother liquor; D) Purification by column chromatography; E) AcetophenoneO-acetyl oxime (3) obtained by column chromatography (photos provided by checkers)
C. 2-Phenyl-4,6-bis(trifluoromethyl)pyridine (5). AcetophenoneO-acetyl oxime (3) (2.12 g, 12.0 mmol, 1.0 equiv) in a 100 mL single-necked (15/25) round-bottomed flask is mixed with toluene (10 mL), which is removed by rotary evaporation under reduced pressure (40 ℃, 16 mmHg) for 10 min. This process is repeated two more times with toluene (2 × 10 mL). An oven-dried football-shaped Teflon-coated magnetic stir bar (10 × 25 mm), NH4I (1.74 g, 12.0 mmol, 1.0 equiv) (Note 19), Na2S2O4 (2.46 g, 12.0 mmol, 1.0 equiv) (Note 20), and powdered molecular sieves 4A (1.06 g) (Note 21) are successively added to the flask. Then, hexafluoroacetylacetone (4) (3.40 mL, 24.3 mmol, 2.0 equiv) (Note 22) and anhydrous toluene (40.0 mL) (Note 23) are added by a plastic syringe (Figure 6A). The flask is capped with a 15/25 three-way stopper with joint Teflon plug, which is connected to a Schlenk line. The juncture of the glassware is sealed with Teflon tape and the stopper is fixed with rubber bands. The flask is evacuated and backfilled with argon three times (Note 24) (Figure 6B). Then, the flask is sealed and disconnected from the Schlenk line. The resulting red solution is placed behind a blast shield and then onto a pre-heated silicon oil bath (130 ℃, oil bath temperature) (Notes 25 and 26) (Figure 6C). After the reaction mixture is stirred for 10 h in the oil bath, the reaction vessel is removed from the oil bath and allowed to cool to 24 ℃ (Note 27) (Figure 6D).

Figure 6. A) Reaction mixture after the addition of toluene; B) Set-up for substituting air with argon in the flask C) Reaction setup; D) Reaction mixture after 10 h (photos provided by checkers)
The resulting solution is filtered through a short pad of silica gel (Note 28) (Figure 7A) and eluted with ethyl acetate (200 mL) into 500 mL round-bottomed flask. The resulting solution is concentrated by a rotary evaporator under reduced pressure (37 ℃, 26 mmHg) for 5 min to afford the crude mixture as a red oil (Figure 7B). The crude residue is purified by flash column chromatography on silica gel using hexane and ethyl acetate as eluents (Note 29) (Figure 7C) to afford 2-phenyl-4,6-bis(trifluoromethyl)pyridine (5) as a light brown oil (2.41 g, 8.40 mmol, 69% yield, 97.7 % purity) (Notes 30, 31, and 32) (Figure 7D).
Figure 7. A) Filtration through a short pad of silica gel; B) Crude red oil; C) Purification by column chromatography; D) 2-Phenyl-4,6-bis(trifluoromethyl)pyridine (5) after column chromatography (photos provided by checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudentpractices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
acetophenone,
hydroxylamine hydrochloride,
anhydrous sodium acetate,
methanol,
ethyl acetate,
acetic anhydride,
hexane,
toluene,
ammonium iodide,
sodium dithionite,
hexafluoroacetylacetone, and molecular sieves 4A.
2. The submitters purchased
hydroxylamine hydrochloride (98.5%) from Hunan Huihong Chemical Reagent Company Limited and used it as received.
Hydroxylamine hydrochloride (99%) was purchased from Sigma-Aldrich and used as received (checkers).
3. The submitters purchased
anhydrous sodium acetate (99%) from Aladdin Chemical Reagent Company Limited and used the material as received.
Anhydrous sodium acetate (99%) was purchased from Thermo Fisher Scientific Co., Inc. and used as received (checkers).
4. The submitters purchased
acetophenone (99%) from Energy Chemical and used the material as received.
Acetophenone (99%) was purchased from Kanto Chemical Co., Inc. and used as received (checkers).
5. The submitters purchased anhydrous
methanol from Tianjin Fuyu Chemical Reagent Company Limited and used the material as received. Anhydrous
methanol (≥99.8%) was purchased from FUJIFILM Wako Pure Chemical Corporation and used as received (checkers).
6. Condenser (C629300) was purchased from Synthware (submitters). Dimroth condenser (No. 1220030) was purchased from Yazawa-Kagaku Co., Inc (checkers).
7. A 500 mL oil bath was used with a stir rate of ~ 500 rpm (submitters). A 650 mL oil bath was used with a stir rate of 800 rpm (checkers).
8. A milky color solution was observed with no color change over the course of the reaction.
9. The reaction was monitored by TLC analysis on silica using
hexane/
ethyl acetate (10/1, v/v) (Figure 8). R
f of
1 = 0.39, R
f of
2 = 0.30.
Figure 8. Thin-layer chromatography (TLC) analysis of the reaction mixture; A) Visualized by 254 nm UV. Left-to-right: Compound 1; Co-spot of compound 1 and the reaction mixture; The reaction mixture; (B) Visualized by anisaldehyde stain. Left-to-right: Compound 1; Co-spot of compound 1 and the reaction mixture; The reaction mixture (photos provided by checkers)
10.
Ethyl acetate (>99.0%) was purchased from Kanto Chemical Co., Inc. and used as received.
11. Characterization data of the non-purified
acetophenone oxime (
2): white solid. mp 53-55 ℃.
1H NMR
pdf (400 MHz, CDCl
3) δ: 7.64-7.62 (m, 2H), 7.39-7.37 (m, 3H), 2.30 (s, 3H).
13C NMR
pdf (100 MHz, CDCl
3) δ: 156.0, 136.4, 129.3, 128.5, 126.0, 12.4. HRMS (ESI
-)
m/z calcd. for C
8H
8NO [M-H]
-134.0611; found 134.0608. A reaction on half-scale provided 1.68 g of the unpurified product.
12. The submitters purchased
acetic anhydride (98.5%) from Chengdu Kelon Chemical Reagent Factory and used the material as received.
Acetic anhydride (>99.0%) was purchased by the checkers from Tokyo Chemical Industry Co., Ltd. and used as received.
13. A 650 mL oil bath was used with a stir rate of 800 rpm. The submitters noted that a light yellow solution was obtained, and no color change was observed over the course of the reaction.
14. The checkers monitored the reaction by TLC analysis on silica using
hexane/
ethyl acetate (5/1, v/v) (Figure 9). R
f of
2 = 0.40, R
f of
3 = 0.29.
Figure 9. Thin-layer chromatography (TLC) analysis of the reaction mixture; A) Visualized by 254 nm UV. Left-to-right: crude 2; Co-spot of crude 2 and the reaction mixture; The reaction mixture; (B) Visualized by anisaldehyde stain. Left-to-right: crude 2; crude 2 and the reaction mixture; reaction mixture (photos provided by checkers)
15. The checkers performed recrystallization of the crude oxime acetate as follows.
Ethyl acetate (2 mL) and
hexane (10 mL) are added to the crude residue at 23 ℃ and the solid is dissolved at 38 ℃. The solution is cooled to 23 ℃ for 8 min and then placed in an ice bath for 2 h to induce the formation of white solid. The solid is vacuum filtered through a Kiriyama-funnel (S-60, fiter paper: No.5B, 60 mmΦ, washed with ice-cooled
hexane (15 mL ×2), and dried under reduced pressure (23 ℃, 4.7 mmHg) for 1 h. 2.51 g of
acetophenoneO-acetyl oxime (
3) is obtained. The purity of compound
3 was determined to be 99.5% by quantitative
1H NMR spectroscopy in CDCl
3 using 15.3 mg of the compound
3 and 15.4 mg of
dibromomethane (Nacalai Tesque, Inc., >99.0%) as an internal standard.
16. Sand was purchased from Nacalai Tesque, Inc. and used as received. Silica gel (Silica gel 60 N, 0.040-0.050 mm, spherical and neutral) was purchased from Kanto Chemical Co., Inc. and used as received.
Hexane (>96.0%) was purchased from Kanto Chemical Co., Inc. and used as received.
Toluene (99%) was purchased from FUJIFILM Wako Pure Chemical Corporation and used as received (checkers). Column chromatography was performed as follows. Flash column (4.8 cm diameter) is charged with sea sand to a height of 2 cm and then with silica gel (120 g) using a wet-pack method to give a column of 15 cm height. Sea sand with a 1 cm height is added to the top of the column. The crude oil is loaded onto the column using 15 mL of
toluene. Fraction collection (100 mL) is then started using test tubes. Elution is continued with 500 mL of
hexane/
ethyl acetate (20/1, v/v), 500 mL of
hexane/
ethyl acetate (15/1, v/v), 1500 mL of
hexane/
ethyl acetate (10/1, v/v), and then 1500 mL of
hexane/
ethyl acetate (6/1, v/v) using compressed air. As illustrated in Figure 10, the fractions containing the
acetophenoneO-acetyl oxime (
3) are identified as fractions No. 18 through No. 24 by TLC (R
f of 0.23,
hexane/
ethyl acetate = 5/1 (v/v), visualized by 254 nm UV). These fractions are combined, concentrated using a rotary evaporator under reduced pressure (37 ℃, 26 mmHg) for 20 min, and then placed under reduced pressure (23 ℃, 6.8 mmHg) for 2 hours to remove residual solvent. Compound 3 (791 mg) is obtained.
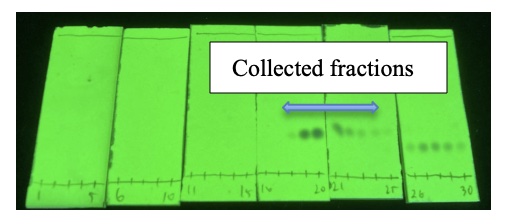
Figure 10. TLC analysis of column chromatography visualized by 254 nm UV (photo provided by checkers)
17. Characterization data of
acetophenoneO-acetyl oxime (
3): white solid, mp 54-56 ℃.
1H NMR
pdf (400 MHz, CDCl
3) δ: 7.75-7.73 (m, 2H), 7.47-7.39 (m, 3H), 2.40 (s, 3H), 2.28 (s, 3H).
13C NMR
pdf (100 MHz, CDCl
3) δ: 168.9, 162.4, 134.8, 130.5, 128.5, 126.9, 19.8, 14.4. IR (KBr film): 3057, 1761, 1616, 1444, 1362, 1310, 1208, 896, 773, 695 cm
−1. HRMS (ESI+)
m/z calcd. for C
10H
11NO
2Na [M+Na]
+ 200.0682; found 200.0685. The purity of the
acetophenoneO-acetyl oxime (
3) was determined to be 99.5% by quantitative
1H NMR
pdf spectroscopy in CDCl
3 using 14.6 mg of the compound
3 and 15.2 mg of
dibromomethane (Nacalai Tesque, Inc., >99.0%) as an internal standard.
18. When the two-step reaction sequence was carried out with 10.3 mmol of
acetophenone (
1),
acetophenoneO-acetyl oxime (
3) was obtained (1.72 g, 94% yield over 2 steps from compound
1, 98.7% purity).
19. The submitters purchased
ammonium iodide (99%) from Bide Pharmatech Ltd, Ih was used as received.
Ammonium iodide (≥99.5%) was purchased from FUJIFILM Wako Pure Chemical Corporation and dried under reduced pressure (4.6 mmHg) for 8 h before use (checkers).
20. The submitters purchased
sodium dithionite (90%) from Aladdin Chemical Reagent Company Limited, Ih was used as received.
Sodium dithionite (>85.0%) was purchased from Tokyo Chemical Industry Co., Ltd. and dried under reduced pressure (4.6 mmHg) for 8 h before use (checkers).
21. Powdered molecular sieves 4A were purchased from Sigma-Aldrich and dried by heating with a heat gun under reduced pressure (4.8 mmHg) for 3 min before use. The checkers found that molecular sieves 4A prevented the hydrolysis of
3 into
acetophenone (
1) under the reaction conditions.
22. The submitters purchased
hexafluoroacetylacetone (98%) from Energy Chemical Reagent Company LIed, which was used as received.
Hexafluoroacetylacetone (>95.0%) was purchased from Tokyo Chemical Industry Co., Ltd. and dried with molecular sieves 4A (beads) purchased from Sigma-Aldrich (checkers).
23. The submitters purchased
toluene from Tianjin Fuyu Chemical Reagent Company Limited and used the solvent as received. Anhydrous
toluene (99.5%) was purchased from FUJIFILM Wako Pure Chemical Corporation and used as received (checkers).
24. The flask, fitted with a three-way stopper, was evacuated by vacuum pump until bubbling of the solution mixture was observed, at which time the flask was backfilled with argon. This process was repeated three times.
25. A 1200 mL oil bath was used with a stir rate of 800 rpm. A light brown solution was quickly formed, after which no color change was observed over the course of the reaction.
26. Because the flask is sealed and then heated, a blast shield should be used to shield the surroundings while heating the reaction mixture.
27. The checkers monitored the reaction by TLC analysis on silica using
hexane/
ethyl acetate (5/1, v/v) (Figure 11). R
f of
3 = 0.30, R
f of
5 = 0.75.
Figure 11. Thin-layer chromatography (TLC) analysis of the reaction mixture visualized by 254 nm UV. Left-to-right: compound 3; co-spot of compound 3 and the reaction mixture; reaction mixture (photo provided by checkers)
28. Silica gel (Silica gel 60 N, 0.100-0.210 mm, spherical and neutral) was purchased from Kanto Chemical Co., Inc. and used as received.
29. Column chromatography was performed as follows. A flash column (5.2 cm diameter) is charged with sea sand to a height of 2 cm and then with silica gel (Silica gel 60 N, 0.040-0.050 mm, spherical and neutral, 100 g) using a wet-pack method to give a column of 11 cm height. Sea sand with a 2 cm height is added to the top of the column. The crude oil is loaded onto the column using 50 mL of
hexane. Fraction collection (100 mL) is then started using test tubes. Elution is continued with 800 mL of
hexane and then 1700 mL of
hexane/
ethyl acetate (100/1, v/v) using compressed air. As illustrated in Figure 12, the fractions containing the
2-phenyl-4,6-bis(trifluoromethyl)pyridine (
5) are identified as fractions No. 7 through No. 22 by TLC (R
f of 0.75,
hexane/
ethyl acetate = 5/1 (v/v), visualized by 254 nm UV). These fractions are combined, concentrated using a rotary evaporator under reduced pressure (37 ℃, 26 mmHg) for 5 min, and then placed under reduced pressure (23 ℃, 6.5 mmHg) for 20 min to remove residual solvent. Compound
5 (2.41 g) is obtained.

Figure 12. TLC analysis of column chromatography visualized by 254 nm UV (photo provided by checkers)
30. Characterization data of
2-phenyl-4,6-bis(trifluoromethyl)pyridine (
5): light brown oil.
1H NMR
pdf (400 MHz, CDCl
3) δ: 8.13-8.10 (m, 3H), 7.80 (s, 1H), 7.56-7.52 (m, 3H).
13C NMR
pdf (100 MHz, CDCl
3) δ: 159.4, 149.5 (q,
JC-F = 35.9 Hz), 140.8 (q,
JC-F = 34.5 Hz), 136.3, 130.7, 129.1, 127.2, 122.4 (q,
JC-F = 275.0 Hz), 121.0 (q,
JC-F = 275.9 Hz), 118.4, 114.3. IR (KBr film): 3072, 1620, 1588, 1462, 1389, 1280, 1199, 1145, 892, 688 cm
−1. HRMS (APCI+)
m/z calcd. for C
13H
8F
6N [M+H]
+ 292.0555; found 292.0556.
31. The purity of the
2-phenyl-4,6-bis(trifluoromethyl)pyridine (
5) was determined to be 97.7% by quantitative
1H NMR
pdf spectroscopy in CDCl
3 using 19.7 mg of the product
5 and 11.6 mg of
dibromomethane (Nacalai Tesque, Inc., >99.0%) as an internal standard (checkers).
32. When the reaction was carried out with 5.98 mmol of
acetophenoneO-acetyl oxime (
3), 1.27 g of
2-phenyl-4,6-bis(trifluoromethyl)pyridine (
5) was obtained (73% yield, 97.6% purity).
3. Discussion
Table 1. NH4I/Na2S2O4-mediated reductive formation of 4,6-bis(trifluoromethyl)pyridines
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved