Org. Synth. 2023, 100, 327-346
DOI: 10.15227/orgsyn.100.0327
Stereoselective Synthesis of (E)-Vinylstannanes from Alkynes via Hydroboration and Transmetalation
Submitted by Ruiyang Guo and Rick L. Danheiser*
1Checked by Stanna K. Dorn and Sarah E. Reisman
1. Procedure (Note 1)
A. 1-((But-3-yn-1-yloxy)methyl)-4-methoxybenzene (1). A 100-mL three-necked, round-bottomed flask (Note 2) is equipped with a 16 x 7 mm Teflon-coated, oval magnetic stir bar, a rubber septum, a glass stopper, and a nitrogen inlet adapter (Note 3). The flask is charged with THF (15 mL) (Note 4), DMF (15 mL) (Note 5) and cooled to -11 ℃ (internal temperature) using an ice-brine bath (Note 6). The rubber septum is pierced with a thermocouple probe and the flask is then charged with 3-butyn-1-ol (1.14 mL, 1.05 g, 15.0 mmol, 1.00 equiv) (Note 7) and 1-(chloromethyl)-4-methoxybenzene (2.24 mL, 2.58 g, 16.5 mmol, 1.10 equiv) (Note 8) via 1-mL disposable syringes (Figure 1). Sodium hydride (NaH) (0.608 g, 60% dispersion in mineral oil, 15.2 mmol, 1.01 equiv) is added in one portion (bubbling and a slight exotherm (~3-4 ℃) is observed upon addition) (Note 9).
Figure 1. Reaction assembly for step A (photo provided by submitter)
The reaction mixture is maintained at -8 ℃ (internal temperature) for 1 h and then the ice-brine bath is replaced with an ice-water bath (internal temperature: 2 ℃). After 3 h, the reaction mixture is quenched (Note 10) by slowly pouring the mixture into 40 mL of stirred ice-cold, saturated aq NH4Cl solution (Note 11) over 3 min. The resulting mixture is transferred to a 500-mL separatory funnel and extracted with ethyl acetate (3 x 30 mL) (Note 12). The combined organic layers are washed with water (5 x 50 mL) and saturated aq NaCl solution (40 mL) (Note 11), dried over 2 g of MgSO4 (Note 11), and filtered through a 60-mL sintered glass Büchner funnel (medium porosity) with the aid of 15 mL of ethyl acetate. The filtrate is concentrated by rotary evaporation (25 ℃, 20 mmHg) to afford 3.55 g of a clear, dark yellow oil.
This oil is dissolved in 50 mL of dichloromethane (Note 13) and concentrated by rotary evaporation onto 7 g of silica gel (Note 14). The resulting material is loaded onto the top of a chromatography column (65 mm diameter) containing 100 g of silica gel prepared as a slurry in hexanes. Elution with 1:20 ethyl acetate-hexanes (Note 15) affords the product in fractions 64-95 (Note 16). These fractions are combined and concentrated by rotary evaporation (25 ℃, 20 mmHg and then 0.1 mmHg) to provide 2.20 g (77%) of 1 as a clear, colorless oil (Notes 17, 18, and 19) (Figure 2).
Figure 2. Butynol PMB ether 1. (a) isolated material (photo provided by submitter);(b) isolated material (photo provided by checker)
B. (E)-Tributyl(4-((4-methoxybenzyl)oxy)but-1-en-1-yl)stannane (2). A 100-mL, three-necked, round-bottomed flask (Note 2) equipped with a glass stopper, a rubber septum fitted with a thermocouple probe, a nitrogen inlet adapter (Note 3), and a 16 x 7 mm Teflon coated oval magnetic stir bar (Figure 3) is charged with cyclohexene (2.13 mL, 1.73 g, 21.0 mmol, 2.0 equiv) (Note 20) via a 1-mL disposable syringe and then 25 mL of THF (Note 4).
Figure 3. Reaction assembly for step B (photo provided by submitter)
The solution is cooled in an ice-water bath and borane-dimethyl sulfide complex (1.00 mL, 0.798 g, 10.5 mmol, 1.0 equiv) (Note 21) is added dropwise over 2 min via a 1-mL disposable syringe while the internal temperature is maintained between 2 and 5 ℃. The colorless solution is stirred in the ice bath for 3 h during which time a white precipitate appears (Figure 4a). A solution of alkyne 1 (2.00 g, 10.5 mmol, 1.00 equiv) in 5 mL of THF is then added dropwise over 2 min via a 5-mL disposable syringe (Note 22), and the resulting white suspension is stirred in the ice bath for 3 h during which time all of the solid goes into solution (Figure 4b). Galvinoxyl (0.0165 g, 0.039 mmol, 0.0037 equiv) (Note 23) is added in one portion by temporarily removing the glass stopper from the neck of the flask, leading to an amber solution. A solution of Bu3SnOMe (3.02 mL, 3.37 g, 10.5 mmol, 1.00 equiv) (Note 24) in 5 mL of THF is then added dropwise over 2 min via a 5-mL disposable syringe (Note 25), and the resulting yellow solution (Figure 4c) is stirred in the ice bath for 2 h (Note 26). Aqueous 10% NaOH solution (15 mL) (Note 27) is added in a rapid dropwise fashion via a 24-mL disposable syringe, and the solution immediately turns red and then purple upon addition of base (Note 28). At this point 30% aqueous H2O2 solution (15 mL) (Note 29) is added dropwise via a 24-mL disposable syringe. Addition of the H2O2 solution results in a significant exotherm so the rate of addition is carefully controlled such that the internal temperature never exceeds 8 ℃, with extra care being taken for the first 2 mL of reagent added (Note 30). The ice bath is then removed, and the reaction mixture is allowed to warm to 25 ℃ and stirred for an additional 30 min (Figure 4d).

Figure 4. Appearance of reaction mixture during the course of the reaction. (a) After addition of BH3⋅SMe2 and stirring for 2 h; (b) after addition of alkyne 1; (c) after addition of Bu3SnOMe; (d) after addition of NaOH and H2O2 (photo provided by submitter)
The two-phase purple reaction mixture is transferred to a 500-mL separatory funnel and extracted with 40 mL of hexanes. The now colorless aqueous layer is extracted with hexanes (2 x 40 mL) (Figure 5), and the combined organic phases are washed with 50 mL of saturated aq NaCl solution, dried over 8 g of MgSO4, and filtered through a 60-mL sintered glass Büchner funnel (medium porosity) with the aid of 30 mL of hexanes. The filtrate is concentrated by rotary evaporation (25 ℃, 20 mmHg and then 0.1 mmHg) (Note 31) to afford 7.50 g of a clear, yellow-orange oil.
Figure 5. Appearance during workup; (a) Upon pouring into funnel; (b) upon addition of hexanes (photo provided by submitter)
This material is dissolved in a minimal volume of hexanes and loaded onto a column (55 mm diameter) containing 150 g of basic alumina (Note 32). Elution with a gradient of 0:100 to 40:60 methyl t-butyl ether-hexanes (Note 33) affords the product in fractions 20-49 (Note 34). These fractions are combined and concentrated by rotary evaporation (25 ℃, 20 mmHg and then 0.1 mmHg, Note 31) to provide 3.20 g (63%) of vinylstannane 2 as a pale yellow oil (Notes 35, 36, 37, and 38) (Figure 6).
Figure 6. (E)-Vinylstannane 2 isolated material (photo provided by checker)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
3-butyn-1-ol,
sodium hydride,
tetrahydrofuran,
1-(chloromethyl)-4-methoxybenzene,
dimethylformamide,
borane-dimethyl sulfide complex,
cyclohexene,
galvinoxyl,
tributyltin methoxide,
hexanes,
dichloromethane and
ethyl acetate.
Tributyltin methoxide and other tributyltin derivatives are moderately toxic. These reagents should only be handled by individuals trained in their proper and safe use.
2. Glassware was flame-dried by the checkers under vacuum (0.2 mmHg), back-filled with argon while hot, and then maintained under inert atmosphere during the course of the reaction. Alternatively, glassware was oven-dried, evacuated on a Schlenk line (0.2 mmHg) / refilled with
nitrogen (3x evacuation + refill), and maintained under inert atmosphere during the course of the reaction.
3. The submitters used an argon atmosphere, while the checkers used a
nitrogen atmosphere.
4.
Tetrahydrofuran (low water HPLC grade) was purchased from J. T. Baker and purified by pressure filtration through activated alumina prior to use.
5.
Dimethylformamide was purchased from J. T. Baker, purified by pressure filtration through activated alumina, and stored for at least 12 h over 3 A molecular sieves before use. In the checker's hands, the storage over molecular sieves was not necessary.
6. The bath is prepared in a 150 x 75 mm crystallizing dish (Figure 6) using 200 g of
NaCl and 600 g of ice. The submitters recorded an internal temperature of -8 ℃.
Figure 7. Ice-brine bath (photo provided by submitter)
7.
3-Butyn-1-ol (98%) was purchased from Oakwood Chemical and used as received.
8.
1-(Chloromethyl)-4-methoxybenzene (98%, stabilized with amylene) was purchased from TCI America and used as received. After usage, the headspace of the container was flushed with argon using an argon balloon to mitigate the buildup of
HCl in the container.
9.
Sodium hydride (60% weight dispersion in mineral oil) was purchased from Sigma-Aldrich and was used as received. The
NaH was weighed on folded weighing paper (Figure 7) and added by removing the glass stopper and temporarily increasing the flow of argon (or
nitrogen) through the open neck of the flask. If
NaH was stored in a bottle (5 g item), the headspace of the container was flushed with an argon balloon after usage.
Figure 8. Sodium hydride dispersion on weighing paper (photo provided by submitter)
10. TLC analysis indicates almost complete disappearance of starting material after 2 h (Figure 8a, submitters) and 3 h (Figure 8b, checkers) of stirring in the ice water bath. TLC analysis was performed with silica gel plates (glass backed, purchased from Sorbtech or Merck KGaA) with 10:1
hexanes-
ethyl acetate as the eluent and visualization with
KMnO4 stain (3 g of
KMnO4, 20 g of
K2CO3, and 5 mL of 5%
NaOH dissolved in 300 mL of water) and UV.
Figure 9. TLC analysis of butynol protection after 2 h; (a) after 2 h (photo provided by submitter); (b) after 3 h (photo provided by checker)
11.
NH4Cl (99.5%),
NaCl (99%), and
MgSO4 (99.5%) were purchased from Sigma-Aldrich and used as received.
12.
Ethyl acetate (≥99.7%) was purchased from Sigma-Aldrich and used as received.
13.
Dichloromethane (≥99.8%) was purchased from Sigma-Aldrich and used as received.
14. Silica gel (VWR "Industrial Grade" irregular silica gel 60A, 40-60 μm, 200-300 mesh) was used as received.
15.
Hexanes (mixture of isomers, ≥99.7%) was purchased from Sigma-Aldrich and used as received.
16. Fractions (15 mL) are collected in 16 x 150 mm test tubes. Submitters collected 20 mL fractions in the same size test tubes and isolated product in fractions 27-45 (2.28 g, 80%). On half-scale (with 8 mL fractions in 16 x 100 mm test tubes), checker isolated product in fractions 26-47. The checkers note that the separation for the column is somewhat challenging (Figure 10).
Figure 10. TLC analysis of butynol protection column (half-scale reaction) to illustrate close Rf values (TLC conditions same as listed in Note 10) (photo provided by checker)
17. Analysis by qNMR
pdf, performed with pyrazine (Millipore Sigma, >99%) as an internal standard, indicated that the purity of the product was 97%.
18. The checkers performed a second reaction at half-scale (7.5 mmol scale) and received 1.24 g (87%) of the product with purity of 97%.
19.
1-((But-3-yn-1-yloxy)methyl)-4-methoxybenzene (
1) has the following physical and spectroscopic properties: R
f = 0.4 (10:1
hexanes-
ethyl acetate; glass-backed Sorbtech or Merck KGaA silica gel plate);
1H NMR
pdf (CDCl
3, 400 MHz) δ: 7.30 (d,
J = 8.6 Hz, 2H), 6.91 (d,
J = 8.6 Hz, 2H), 4.52 (s, 2H), 3.83 (s, 3H), 3.60 (t,
J = 6.9 Hz, 2H), 2.51 (dt,
J1 = 2.6 Hz,
J2 = 6.9 Hz, 2H), 2.01 (t,
J = 2.6 Hz, 1H);
13C NMR
pdf (CDCl
3, 101 MHz) δ: 159.3, 130.1, 129.4, 113.8, 81.4, 72.7, 69.3, 67.9, 55.3, 19.9; FTIR (
NaCl, thin film) (cm
-1): 3290, 2935, 2911, 2861, 2836, 1612, 1585, 1511, 1462, 1361, 1301, 1244, 1173, 1092, 1032, 819, 756, 636, 581, 515; HRMS (FD)
m/z calcd for C
12H
14O
2 [M+]: 190.0994. Found: 190.0995.
20.
Cyclohexene was purchased from EM Science or Fisher Scientific (Spectrum). Prior to use, 15 mL of
cyclohexene was transferred to a separatory funnel and washed with 15 mL saturated aq
NaHSO3 solution and then 15 mL of water. The
cyclohexene was next dried over
CaCl2, filtered, and distilled from ca. 2 g of
CaCl2 under an atmosphere of argon using a short-path distillation apparatus. An approximately 2-mL forerun was collected, at which point the temperature measured at the still head stabilized at 75 ℃ (bath temp 100 ℃) and the main fraction was then collected for use (checkers). For submitters, the procedure was done identically but the still head stabilized at 60 ℃ (bath temperature 92 ℃).
21.
Borane-dimethyl sulfide complex was purchased from Oakwood Chemical, stored in a Schlenk flask, and used as received. In the checker's hands, storage in a Schlenk flask was not necessary but the container was handled under an inert atmosphere.
22. A solution of alkyne
1 in 3 mL of
THF was prepared under argon in a 25-mL one-necked, pear-shaped flask fitted with a rubber septum and transferred to the reaction mixture with a 5-mL syringe. The flask was washed with
THF (2 x 1 mL). The internal temperature was 2 to 4 ℃ during the addition.
23.
Galvinoxyl was purchased from Sigma-Aldrich and used as received.
24.
BuÂ3SnOMe (97%) was purchased from Fisher Scientific and used as received.
25. The solution of
Bu3SnOMe in 3 mL of
THF was prepared under argon in a 25-mL one-necked, pear-shaped flask fitted with a rubber septum and transferred to the reaction mixture with a 5-mL syringe. The flask was washed with
THF (2 x 1 mL). The internal temperature was 2 to 6 ℃ during the addition.
26. TLC analysis was performed with silica gel plates (glass backed, purchased from Sorbtech or Merck KGaA) with 10:1
hexanes-
ethyl acetate as the eluent and visualization with
KMnO4 stain (3 g of
KMnO4, 20 g of
K2CO3, and 5 mL of 5%
NaOH dissolved in 300 mL of water) and PMA stain (12 g of phosphomolybdic acid dissolved in 250 mL EtOH) (Figure 11). TLC analysis can also be performed with basic
aluminum oxide glass-backed plates (purchased from Merck KGaA, same eluent/stains as previously listed).
Figure 11. TLC analysis 2 h after addition of Bu3SnOMe (a) silica gel (submitter image) (b) silica gel (checker image) (c) basic alumina (photo provided by checker)
27.
Sodium hydroxide (>98%, pellets) was purchased from Sigma-Aldrich and used as received. A 10% w/v solution was prepared by slowly adding 50 g of
NaOH to 300 mL of water and then adding water to a final volume of 500 mL after the solution had returned to room temperature.
28. The submitters added the
NaOH solution in one portion; however, the checkers added the solution more slowly, albeit in a rapid dropwise fashion, due to observation of a small exotherm (~2-3 ℃) during addition. The total addition time was ~5-10 min.
29. Aqueous
hydrogen peroxide solution (30%) was purchased from Alfa Aesar or from Thermo Scientific (29-32% aqueous solution) and used as received.
30. The submitters note that during peroxide addition, the rate of addition needed to be carefully controlled so that the internal temperature returns to 0 ℃ before additional drops of solution were added; however, the checkers noted that there was a ~0.3-0.5 ℃ exotherm with each drop during initial addition, which required >30 seconds to return to base temperature. To avoid an unnecessarily long addition period, the rate of addition was controlled such that the internal temperature never exceeded 8 ℃. With this adjustment, it took ~1 h to add the first 2 mL of peroxide, after which the exotherm stabilized and the rate of addition could be increased to a rapid dropwise fashion (internal temperature never exceeding 8 ℃).
31. Rotary evaporation (20 mmHg) was performed in a fume hood to properly ventilate tin-containing compounds.
32.
Aluminum oxide (activated, basic, Brockmann I) was purchased from Sigma-Aldrich and used as received. The submitters note that the activity of the alumina varies depending on its age. The conditions described herein are for alumina that ranged from a freshly opened bottle to three months old.
33.
Methyl tert-butyl ether (≥99.0%) was purchased from Sigma-Aldrich and used as received.
34. Fractions (20 mL) are collected in 16 x 150 mm test tubes. The column gradient was increased by 10% (e.g., from 0:100 to 10:90) approximately every 200 mL, eventually eluting with 40:60
MTBE-
hexanes until the product was no longer detected in fractions by TLC. TLC analysis on silica gel plates may show some decomposition on the plate, resulting in a second, faint spot at R
f = 0.4. The submitters note that product elutes in fractions 17-82 under these conditions (2.906 g isolated, 57%); in the checker's hands, product eluted in fractions 20-49.
35. Analysis by qNMR
pdf, performed with pyrazine (Millipore Sigma, >99%) as an internal standard, indicated that the purity of the product was 97%.
36. The checkers performed a second reaction at half-scale (5.25 mmol scale) and received 1.52 g (60%) of the product with purity of 97%.
37.
(E)-Tributyl(4-((4-methoxybenzyl)oxy)but-1-en-1-yl)stannane (
2) has the following physical and spectroscopic properties: R
f = 0.6 (10:1
hexanes-
ethyl acetate; glass-backed Sorbtech or Merck KGaA silica gel plate) R
f = 0.8 (10:1
hexanes-
ethyl acetate; glass-backed Merck KGaA basic alumina plate);
1H NMR
pdf (CDCl
3, 400 MHz) δ: 7.29 (d,
J = 8.7 Hz, 2H), 6.90 (d,
J = 8.7 Hz, 2H), 6.12 - 5.96 (m*, 2H), 4.48 (s, 2H), 3.83 (s, 3H), 3.53 (t,
J = 6.9 Hz, 2H), 2.47 (ttd,
J = 6.9, 4.7, 2.3 Hz, 2H), 1.62 - 1.39 (m*, 6H), 1.39 - 1.26 (m*, 6H), 1.00 - 0.78 (m*, 15H). *additional complex splitting due to
119Sn and
117Sn isotopes.
13C NMR
pdf (CDCl
3, 101 MHz) δ : 159.1, 145.5, 130.7, 130.0, 129.3 (
JSn(119)-C = 391 Hz,
JSn(117)-C = 373 Hz), 113.8, 72.5, 69.5, 55.3, 38.2 (
JSn-C = 61.9 Hz), 29.1 (
JSn-C = 20.2 Hz), 27.3 (
JSn-C = 53.9 Hz), 13.7, 9.4 (
JSn(119)-C = 342 Hz,
JSn(117)-C = 326 Hz); FTIR (
NaCl, thin film) (cm
-1): 2956, 2924, 2851, 1613, 1512, 1463, 1419, 1375, 1358, 1300, 1247, 1171, 1098, 1039, 990, 960, 825, 756, 633; HRMS (FD)
m/z calcd for C
20H
33O
2Sn [M - C
4H
9]: 425.1503. Found: 425.1506.
38.
(E)-Tributyl(4-((4-methoxybenzyl)oxy)but-1-en-1-yl)stannane (
2) is stable to storage neat, under argon at 0 ℃, for at least 6 months.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
(
E)-Vinylstannanes have a number of applications as synthetic intermediates,
2 including most prominently as precursors to vinyllithium compounds and as substrates in Stille coupling reactions.
3 While Suzuki and Negishi coupling reactions are popular alternatives for cross-coupling, in some cases Stille reactions proceed in higher yield, which can be an important consideration in the late stages of total synthesis projects.
One popular general synthetic approach to vinylstannanes
4 involves halogen-tin exchange beginning with vinyl halides which often are prepared from alkynes. Other common routes involve the direct or one-pot conversion of alkynes to vinylstannanes by hydrostannation
4 (radical-mediated, transition-metal-catalyzed, or Lewis-acid-catalyzed), via hydrometalation/transmetalation reactions (involving vinylaluminum, -boron, or -zirconium intermediates), and via the direct addition of trialkylstannyl metal compounds to acetylenes. While many reactions involving these approaches proceed with high stereoselectivity, some suffer from complications due to the formation of stereo- and regioisomers.
Illustrated in this
Organic Syntheses article is a highly stereoselective one-pot procedure based on a method introduced by Hoshi
5 et al. in 2009. Hydroboration of an alkyne with dicyclohexylborane provides an (
E)-vinylborane which upon exposure to tributyltin methoxide undergoes boron to tin transmetalation at room temperature to afford the desired (
E)-vinylstannane in generally good yield and with very high
E/Z stereoselectivity (Scheme 1).
Scheme 1
To demonstrate the Hoshi method, we chose as our substrate the PMB ether derivative of 3-butynol. The detailed procedure described in this article represents the first experimental procedure to appear in
Organic Syntheses for the protection of alcohols with this important protective group.
6A variety of conditions have been reported previously for the synthesis of the PMB ether derivative of 3-butynol, most often involving the alkylation of the butynol with
p-methoxybenzyl chloride,
7 bromide,
8 or iodide.
9 We obtained good results employing the benzylic chloride which is considerably less expensive than the bromide. While prior procedures used either THF or DMF as reaction solvent, we obtained best results with a THF-DMF mixture. Prior reports described using 1.1 to 2.0 equiv of sodium hydride to generate the sodium salt of the alcohol; we found 1.0 equiv of NaH and 1.1 equiv of the benzylic chloride to be optimal. It should be noted that the protection of butynol has also been reported by reaction of the alcohol with the trichloroimidate derivative of
p-methoxybenzyl alcohol
10 in the presence of acid and by the direct reaction of butynol with
p-methoxybenzyl alcohol in the presence of Yb(OTf)
311 or FeCl
3.
12In the course of optimizing the conditions for the protection of 3-butynol we observed the formation of varying amounts of the isomeric allenyl ether 3 as a byproduct that was difficult to separate from the desired alkyne. For example, in the worst case, a 66:34 mixture of alkynyl ether 1 to allenyl ether 3 was obtained when the butynol substrate was added to a suspension of excess (1.2 equiv) NaH in THF-DMF at 2 ℃ followed by subsequent addition of the benzylic chloride. Fortunately, after considerable experimentation, we found that formation of the allene byproduct could be suppressed by adding 1.0 equiv of NaH to a solution of both the alcohol and benzylic chloride maintained at a somewhat lower temperature (-8 ℃) in the same solvent mixture.
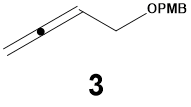
Only one report of the synthesis of (
E)-vinylstannane
2 has previously appeared in the literature. Lee and coworkers carried out hydrostannation of
1 with Bu
3SnH in the presence of AIBN in toluene at 130 ℃ and obtained
2 in 70% yield; the ratio of
E and
Z isomers was not reported.
13 Several laboratories have reported the synthesis of vinylstannanes corresponding to
2 but incorporating different hydroxyl protective groups. For example, in 1985 Oshima described the synthesis of the pure (
E)-vinylstannane benzyl ether in 88% yield via reaction of the corresponding alkyne with 3 equiv of Bu
3SnMgMe in the presence of catalytic CuCN.
14 Oshima subsequently reported hydrostannation of the same acetylene with Bu
3SnH and catalytic Et
3B which afforded the vinylstannane in 71% yield as a 90:10 mixture of
E and
Z isomers. Pattenden described an alternative approach in which the benzyl ether of 3-butynol was first converted to the alkynyl bromide with NBS and AgNO
3 and then the resulting 1-bromoalkyne was treated with 2.2 equiv of Bu
3SnH in the presence of Pd
2(dba)
3-Ph
3P; this produced the desired vinylstannane with >95%
E/Z selectivity.
16 Finally, several reports have appeared on the hydrostannation of the corresponding
t-butyldimethylsilyl ether derivative of 3-butynol; in these reactions the (
E)-vinylstannane is typically accompanied by 8-10% of the
Z isomer.
17We found that the hydroboration/transmetalation procedure described in this article produced the (E)-vinylstannane 2 without the formation of detectable Z isomer provided that the reaction was carried out in the presence of galvinoxyl (0.4 mol%) to inhibit any radical-mediated isomerization of the vinylstannane. In the absence of galvinoxyl, the (E)-vinylstannane was obtained contaminated by a small amount (1%) of the Z isomer.
Finally, it should be noted that we were unable to accomplish the purification of (E)-vinylstannane 2 by silica gel chromatography without observing significant decomposition. Chromatography with basic alumina, on the other hand, gave good separation and provided the desired vinylstannane without the formation of decomposition products that was observed when silica gel was used for purification.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
3-Butyn-1-ol; (927-74-2)
1-(Chloromethyl)-4-methoxybenzene; (824-94-2)
NaH: sodium hydride; (7646-69-7)
Cyclohexene; (110-83-8)
Borane-dimethyl sulfide; (13292-87-0)
Galvinoxyl: (2370-18-5)
BuÂ3SnOMe, tributyltin methoxide: tributylmethoxystannane; (1067-52-3)
NaOH: sodium hydroxide; (1310-73-2)
H2O2: hydrogen peroxide (7722-84-1)

|
Ruiyang (Michelle) Guo is currently an undergraduate at the Massachusetts Institute of Technology expecting to graduate in 2023 with a Bachelor's of Science in Chemistry and Biology. Michelle plans to go on to pursue graduate studies in chemistry. In 2021, Ruiyang joined the Danheiser group as an undergraduate researcher. |

|
Rick Danheiser received his undergraduate education at Columbia where he carried out research in the laboratory of Professor Gilbert Stork. He received his Ph.D. at Harvard in 1978 working under the direction of E. J. Corey on the total synthesis of gibberellic acid. Dr. Danheiser is the A. C. Cope Professor of Chemistry at MIT where his research focuses on the design and invention of new annulation and cycloaddition reactions, and their application in the total synthesis of biologically active compounds. |

|
Dr. Stanna K. Dorn received her B.S. in Chemistry and B.A. in Music from Hope College, where she performed organometallic research with Prof. Jeffrey B. Johnson. In 2022, she obtained her Ph.D. at Indiana University, where she worked on cooperative catalysis methods for alkene difunctionalization under the direction of Prof. M. Kevin Brown. Currently, she is an NIH F32 NRSA Postdoctoral Fellow at Caltech, working under the guidance of Prof. Sarah E. Reisman on the synthesis of complex natural products. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved