Org. Synth. 2023, 100, 347-360
DOI: 10.15227/orgsyn.100.0347
Synthesis of 4-Phenylpyrrolidin-2-one via an Aza-Baeyer-Villiger Rearrangement
Submitted by Mike Ong,
1 Marlene Arnold,
1 and Johannes M. Wahl*
2Checked by Shashwati Paul and M. Kevin Brown
1. Procedure (Note 1)
A. 4-Phenylpyrrolidin-2-one (3). A 500 mL three-necked round-bottomed flask (main and side necks: 29/32 joint) is equipped with a Teflon-coated magnetic stir bar (5 × 1 cm, cylindrical shaped), a 100 mL pressure equalizing addition-funnel open to the air, a thermometer inserted through a thermometer adapter, and a glass stopper. In the flask O-(diphenylphosphinyl)hydroxylamine (2) (DPPH; 8.10 g, 34.7 mmol, 1.16 equiv) (Note 2) is suspended in N,N-dimethylformamide (DMF) (100 mL) (Note 3). The suspension is heated to 25 ℃ (internal temperature) with an outer metal heating block while stirring at approximately 250 rpm (Figure 1). A solution of 3-phenylcyclobutanone (1) (4.39 g, 30.0 mmol, 1.00 equiv) (Note 4) in DMF (60 mL) is added dropwise via the addition-funnel over a period of 15 min. After complete addition, the addition-funnel is replaced by a glass stopper and stirring is continued for 24 h (Note 5) (Figure 2 A-C).

Figure 1. Reaction set-up with reagents before the addition of cyclobutanone solution (photo provided by submitter)
Figure 2. A) reaction appearance after complete addition of cyclobutanone solution; B) reaction appearance after 4 h reaction time; C) reaction appearance after 5 h reaction time (photos provided by submitter)
Figure 3. A) Inverse splash head used for evaporation of DMF; b) Set up for the evaporation of DMF; C) Precipitate formed after 20 min of evaporation (photos provided by submitter)
The reaction mixture is equally portioned between two 250 mL single-necked round-bottomed flasks (14/23 joints) (Notes 6 and 7). The solvent in each flask is removed on a rotary evaporator (50 ℃, 6 mmHg to 3 mmHg) (Note 8) using an inverse splash head (Figure 3 A-C).
The dry fractions are combined in a 250 mL single-necked round-bottomed flask (29/32 joint) using dichloromethane (CH2Cl2) (70 mL) (Note 9) as the transfer solvent. The solvent is removed under reduced pressure using a rotary evaporator (40 ℃, 450 mmHg to 3 mmHg) (Note 10).
The crude material is purified by chromatography on silica gel (Note 11) using ethyl acetate (EtOAc) (Note 12) and methanol (MeOH) (Note 13) as eluents (Note 14) to obtain lactam 3 (3.12 g, 19.4 mmol, 65%, >99% purity) (Notes 15, 16, and 17) as a white solid (Figure 4).
Figure 4. Isolated 4-phenylpyrrolidine-2-one after column chromatography (photo provided by submitter)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
O-(diphenylphosphinyl)hydroxylamine,
3-phenylcyclobutanone,
N,N-dimethylformamide,
dichloromethane,
ethyl acetate,
methanol and silica gel.
2.
O-(Diphenylphosphinyl)hydroxylamine (95%) was purchased from Combi-Blocks (checkers) or from abcr (submitters) and used as received. It can also be obtained by a procedure described by Benkovics and coworkers.
3 Care should be taken when working with
O-(diphenylphosphinyl)hydroxylamine, as this compound contains a decomposition energy of >800 J/g and an onset temperature <100 ℃.
43.
N,N-Dimethylformamide (≥ 99.8%) was purchased from Merck and used as received.
4. The checkers purchased
3-phenylcyclobutanone (98%) (
1) from Ambeed, which was used as received. The submitters prepared
3-phenylcyclobutanone (
1) following a procedure described by Ghosez and coworkers.
5 That procedure was slightly modified using styrene (50.0 g, 54.9 mL, 480 mmol, 4.00 equiv),
N,N-dimethylacetamide (10.5 g, 11.2 mL, 120 mmol, 1.00 equiv),
trifluoromethanesulfonic anhydride (40.6 g, 23.7 mL, 144 mmol, 1.20 equiv.),
2,6-lutidine (15.4 g, 16.7 mL, 144 mmol, 1.20 equiv.),
1,2-dichloroethane (480 mL) and
H2O (64 mL). The crude reaction mixture was purified by column chromatography (
n-pentane:
EtOAc 95:5 to 90:10) followed by bulb-to-bulb distillation (97 ℃ - 125 ℃, 0.13 mmHg). The product (14.8 g, 101 mmol, 85%, 99% purity) was isolated as colorless oil. The purity of compound
1 (>99%) was determined by quantitative qNMR analysis using compound
1 (31.3 mg, 214 μmol) and
1,3,5-trimethoxybenzene (12.0 mg, 71.3 μmol) as an internal standard. The purity was calculated according to the standard method integrating the alpha
CH2 resonance against the aromatic CH signal and showed >99 wt% purity. Styrene (≥99%, Sigma-Aldrich),
N,N-dimethylacetamide (99%, abcr),
trifluoromethanesulfonic anhydride (98%, Carbolution),
2,6-lutidine (>98%, TCI) and
1,2-dichloroethane (≥99%, Carl Roth) were purchased and used as received.
n-Pentane (95%) was purchased from Titolchimica and distilled before use.
5. The progress of the reaction was monitored via TLC analysis with
EtOAc:
MeOH (95:5) used as the eluent. TLC silica gel 60G F254 pre-coated glass backed plates (0.25 mm) were purchased from Merck. The plate was visualized using 275 nm UV-lamp and
KMnO4-stain. Product (
3) R
f = 0.46, starting materials:
cyclobutanone 1 R
f = 0.98,
DPPH (
2) R
f = 0.57 and side products:
cyclobutanone oxime ester R
f = 0.73,
diphenylphosphinic acid R
f = 0.00.
Figure 1. TLC analysis of reaction mixture (95:5 EtOAc:MeOH) with S=starting material (1), M=mixture of 1 and reaction mixture, R=reaction mixture, visualized with 275 nm UV-lamp (left) and KMnO4-stain (right) (photos provided by submitter)
6. The reaction mixture was portioned to accelerate the removal of
DMF due to an increased solvent surface. The submitters tried to remove
DMF by aqueous extraction, which led to significantly lower yields due to the water solubility of the lactam product.
7. The reaction flask was rinsed three times with
DMF (50 mL) to achieve a quantitative transfer of the reaction mixture.
8. After 20 min of evacuation (50 ℃, 6 mmHg) a white precipitate was formed. The collecting flask of the rotary evaporator was emptied, and the precipitate was dried under reduced pressure using a rotary evaporator (50 ℃, 3 mmHg) for further 45 min.
9.
Dichloromethane (≥ 99.5%) was purchased from VWR chemicals and used as received.
10. After removal of the solvent (20 min at 40 ℃, 450 mmHg), the flask was evacuated to 3 mmHg at 40 ℃ for 20 min.
11. The checkers used Siliacycle Siliaflash, Irregular Silica Gel P60 40-63 mm 60 purchased from Fisher Scientific. The submitters used Geduran
® Si 60 (particle size 40-63 μm) from Merck.
12.
Ethyl acetate (≥ 99.5%) was purchased from Fisher Scientific and used as received.
13.
Methanol (≥ 99.8 %) was purchased from VWR chemicals and used as received.
14. Silica gel flash column chromatography was performed using compressed air. A column (6.5 cm OD × 29 cm height) was pre-equilibrated with silica gel (350 g) in
EtOAc (900 mL). The crude product was suspended in
CH2Cl2 (70 mL) and silica gel (15.0 g) (
Note 10) was added. The solvent was removed under reduced pressure using a rotary evaporator (40 ℃, 450 mmHg to 75 mmHg) until a fine powder resulted. The product-adsorbed silica was added to the packed column. Sand (0.4-0.8 mm purchased from Carl Roth) was added to the round-bottomed flask and swirled to remove the silica from the side of the flask to ensure quantitative transfer. The column was eluted with
EtOAc (1.4 L), followed by 95:5
EtOAc:
MeOH (2.4 L). The eluting solvent was collected in 70 mL tubes. Starting from fraction 36 the fraction volume was reduced to 35 mL. Fractions containing the product
3 were identified via TLC analysis (R
f = 0.41, 96:4
EtOAc:
MeOH, visualized using
KMnO4-stain) (Figure 6). The desired product eluted in fractions 42-87. The product spots of fraction 42 and 67-87 overlapped with that of impurities. Fractions 43-66 were combined. The tubes were rinsed three times with
EtOAc (9 mL) to achieve a quantitative transfer. The solvent was removed under reduced pressure (40 ℃, 150 mmHg to 6 mmHg) and the residue was dried under high vacuum (25 ℃, 0.04 mmHg) for 2 h to deliver the desired lactam with 90% purity, assessed through the technique described in
Note 16. Next, the impure lactam was purified by Medium-pressure liquid chromatography (MPLC) technique using a Teledyne ISCO CombiFlash Rf 150 instrument to obtain the lactam >99% purity. A 80 gram RediSepR
f High performance GOLD column was used for the purification. The crude product was suspended in
CH2Cl2 (30 mL) and silica gel (10.0 g) was added. The solvent was removed under reduced pressure using a rotary evaporator (40 ℃) until a fine powder resulted. The product-adsorbed silica was packed on a 25 g RediSepR
f solid sample cartridge. The column was eluted with
EtOAc (2 mins, 60 mL/min flow rate), followed by 99:1
EtOAc:
MeOH (35 min, 60 mL/mins), next gradually increased to 95:5
EtOAc:
MeOH (60 mL/mins) over 5 mins and flushed with 90:10
EtOAc:
MeOH (60 mL/mins) for 5 mins. The eluting solvent was collected in 25 mL tubes. Fractions containing the product
3 were identified via TLC analysis (R
f = 0.5, 96:5
EtOAc:
MeOH, visualized using
KMnO4-stain). The desired product eluted in fractions 23-40. The tubes were rinsed three times with
EtOAc (5 mL) to achieve a quantitative transfer. The solvent was removed under reduced pressure and the residue was dried under high vacuum (25 ℃, 0.4 mmHg) for 30 min.

Figure 2. TLC analysis of column chromatography (96:4 EtOAc:MeOH) visualized with KMnO4-stain (photos provided by submitter)
15.
4-Phenylpyrrolidin-2-one (
3) has the following physical and spectroscopic properties: mp: 74-77 ℃.
1H NMR
pdf (600 MHz, CDCl
3) δ: 7.36-7.32 (m, 2H, C
Harom.), 7.29-7.23 (m, 3H, C
Harom.), 7.12-7.01 (br s, 1H, N
H), 3.79 (dd,
J = 9.6, 8.2 Hz, 1H, NC
HH), 3.69 (
app. p,
J ≈ 8.4 Hz, 1H, PhC
H), 3.43 (dd,
J = 9.6, 7.4 Hz, 1H, NCH
H), 2.74 (dd,
J = 16.9, 9.0 Hz, 1H, COC
HH), 2.51 (dd,
J = 16.9, 8.9 Hz, 1H, COCH
H).
13C NMR
pdf (151 MHz, CDCl
3) δ: 178.1 (
Cq), 142.2 (
Cq), 128.9 (
CH), 127.2 (
CH), 126.9 (
CH), 49.7 (N
CH
2), 40.4 (
CH), 38.2 (CO
CH
2). IR (neat):
ṽ = 3236 (br), 2877 (br), 1693 (s), 1494 (m), 1454 (w), 1360 (w), 1290 (w), 1258 (m), 1050 (w), 759 (m), 700 (m), 472 (w) cm
-1. HRMS (ESI) (
m/z): calculated for C
10H
12NO [M+H]
+: 162.0913, found: 162.0911.
16. The purity of compound
3 (>99%) was determined by quantitative
1H NMR
pdf analysis with the relaxation delay set to 30 sec using 34.6 mg (0.214 mmol) of compound
3 and 13.3 mg (0.079 mmol) of
1,3,5-trimethoxybenzene (BLD pharm, >99.9%, used as received) as an internal standard.
17. When the reaction was carried out on a 10.0 mmol scale, compound
3 (1.30 g, 8.09 mmol, 81%) was obtained with 98.6% purity.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The synthesis of γ-lactams represents a critical area of current research, because these compounds have been identified as promising scaffolds for the development of new drugs due to their diverse and often potent biological activities.
6,7,8 Facile access to γ-lactam analogues provides a platform to explore structure activity relationships enabling the development of novel therapeutic agents.
Insertion of a nitrogen group in the α-position of a carbonyl is well established by the Schmidt and Beckmann reactions. However, these procedures often entail harsh reaction conditions rendering them less suitable for late-stage transformations and lowering the functional group compatibility. To address these issues, we have recently found that DPPH (
2) can act as a simple and commercially available nitrogen source for the ring-expansion of cyclobutanones.
9 While the reagent is typically known for electrophilic aminations, it reveals advantageous properties for nitrogen insertions due to the favorable leaving group present at the nitrogen atom of the reagent. A mechanistic investigation unveiled rearrangement from a Criegee-like intermediate (
4) via an aza-Baeyer-Villiger type reaction, a decisive contrast to the venerable Beckmann rearrangement (Scheme 1). This deduction is based on the observation that oxime ester
5 was formed as an unintended side-product that could not be converted to the γ-lactam under the reaction conditions.
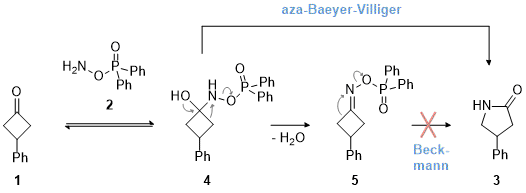
Scheme 1. Mechanistic proposal for the aza-Baeyer-Villiger reaction
The highlights of the methodology can be reflected in the ability of the method to accommodate a wide range of functional groups with minimal interference. In addition, our laboratory has recently disclosed highly regioselective (82:18 to >99:1 regioisomeric ratio (rr)) and stereospecific (>99% enantiospecificity (es)) nitrogen insertions using DPPH (
2), as showcased by the synthesis of γ-lactam
7 (Scheme 2, top).
9 The successful synthesis of rolipram (
9) and its
N-alkylated analogues (
10 and
11) (Scheme 2, bottom) further demonstrates the potential for precise optimization of lead structures, emphasizing the method's suitability for "late-stage skeletal editing".
9Scheme 2. Top: Regioselectivity and enantiospecificity of the aza-Baeyer-Villiger reaction. Bottom: Synthesis of rolipram and its N-alkylated analogues by "late-stage skeletal editing"
Appendix
Chemical Abstracts Nomenclature (Registry Number)
DPPH: O-(Diphenylphosphinyl)hydroxylamine; (2) (72804-96-7)
DMF: N,N-Dimethylformamide; (68-12-2)
3-Phenylcyclobutanone; (1) (52784-31-3)
CH2Cl2: Dichloromethane; (75-09-2)
EtOAc: Ethyl acetate; (141-78-6)
MeOH: Methanol; (67-56-1)

|
Marlene Arnold received her B.Sc. and M.Sc. in chemistry from Ludwig Maximilian University of Munich. She finished her M.Sc. studies with a research stay in the group of Prof. Helma Wennemers (ETH, Zurich), working on peptide catalysis. In 2021, she started her Ph.D. studies at Johannes Gutenberg University Mainz in Prof. Johannes Wahl's laboratory. Her current research focuses on developing new enantioselective reactions with strained rings. |

|
Mike Ong obtained his B.Sc. in chemistry from WWU Münster working on C-H activation of aliphatic carboxylic acids in the group of van Gemmeren. After a research stay in the Rovis lab at Columbia University, he joined the Wahl laboratory in 2020 and earned his M.Sc. degree under the same supervision. Currently, he is pursuing his PhD studies in the same research group, with a particular emphasis on the nitrogen insertion in prochiral cyclobutanones. |

|
Johannes M. Wahl attended the Universität Basel, Switzerland, where he obtained his M.Sc. in 2012. After a research stay at the Scripps Research Institute (Prof. D. G. Blackmond), he moved to Munich to pursue his Ph.D. at the Technische Universität with Prof. T. Bach. After a postdoctoral stay at Indiana University (Prof. M. K. Brown), he started his independent career at the Westfälische-Wilhelms Universität Münster. Since 2020, he holds an Assistant Professorship at the Johannes Gutenberg-Universität in Mainz. |

|
Shashwati Paul was born in Chandernagore, West Bengal, India. She received her Bachelor of Science (B.Sc.) in Chemistry honors from Chandernagore College and Master of Science (M.Sc.) from Indian Institute of Science, Bangalore. Upon completion of her master's study, she moved to Indiana in 2019 to pursue a Ph.D. degree under the supervision of Prof. M. Kevin Brown at Indiana University Bloomington. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved