1. Procedure (Note 1)
A. (E)-Di-tert-butyl(hex-2-en-1-yloxy)silanol (1). An oven-dried, single-necked (24/40 joint) 500 mL round-bottomed flask is equipped with a Teflon-coated magnetic stir bar (4.0 cm x 1.0 cm, oval) and septum. The vessel is purged with argon from a Schlenk line using a 20G inlet and a 20G exit needle connected to an oil bubbler for 30 min (Figure 1A) (Note 2). The flask is then charged with di-tert-butylsilyl bis(trifluoromethanesulfonate) (20.4 mL, 62.6 mmol, 1.25 equiv) (Note 3), using a plastic syringe fitted with a 20G needle, and CH2Cl2 (200 mL) (Note 4), using a plastic syringe fitted with a 20G needle. The reaction flask is then placed in an ice-water bath. After ten minutes, 2,6-lutidine (14.6 mL, 125 mmol, 2.5 equiv) (Note 5) is added dropwise over 10 min using a plastic syringe fitted with a 20G needle. After the reaction mixture is stirred (250 rpm) for 15 min at 0 ℃, trans-2-hexen-1-ol (5.9 mL, 50 mmol, 1 equiv) (Note 6) is then added dropwise using a plastic syringe fitted with 20G needle over 10 min, and the solution is stirred (250 rpm) at 0 ℃ for 1 h (Figure 1B). The progress of the reaction is monitored by thin-layer chromatography (TLC) (Notes 7 and 8) and trans-2-hexen-1-ol is consumed approximately after 1 h following addition (Figure 2). The reaction is quenched at 0 ℃ by careful, portion-wise addition of saturated aqueous NaHCO3 solution (50 mL in 3 portions) (Note 9). The reaction mixture is then transferred to a 1 L separatory funnel. The organic layer is separated and washed with additional portions of saturated, aqueous NaHCO3 solution (3 x 75 mL). The organic layer is then washed with 1 M aqueous HCl solution (2 x 75 mL) (Note 10). The organic layer is separated and dried over anhydrous MgSO4 (5 g) (Note 11), filtered under house vacuum (~75 mm Hg) through a 60 mL sintered glass funnel (50 mm frit, coarse) into a 500 mL round-bottomed flask, then concentrated under reduced pressure using a rotary evaporator (Note 12). The resulting residue (Figure 3A) is purified by flash column chromatography on silica gel (Notes 13, 14, 15 and 16). The fractions containing the product (Figure 3B) are combined and concentrated under reduced pressure on a rotary evaporator (Note 17) followed by high vacuum (20 ℃, <1 mmHg) for 2 h, to give 1 as a colorless oil (8.67 g, 33.5 mmol, 67%) (Figure 4) (Notes 18 and 19).

Figure 1. Reaction set-up for Step 1. (A) Reaction flask purging under argon (B) After addition of 2,6-lutidine and trans-2-hexen-1-ol to di-tert-butylsilyl bis(trifluoromethanesulfonate) in CH2Cl2 at 0 ℃. (Photos provided by checkers)
Figure 2. TLC of the crude reaction mixture, stained with ceric ammonium molybdate stain. Mobile phase: 10 % EtOAc in hexanes. Note: S = starting material, C = co-spot lane, R = reaction mixture. Starting material trans-2-hexen-1-ol (Rf = 0.17), product 1 (Rf = 0.54) (Photo provided by checkers)
Figure 3. (A) Product before purification. (B) Compound 1 is found in fractions 46-81 (Photos provided by checkers)
Figure 4. (E)-Di-tert-butyl(hex-2-en-1-yloxy)silanol (1) is a colorless oil (Photo provided by checkers)
B. Di-tert-butyl(((2R*,3R*)-3-propyloxiran-2-yl)methoxy)silanol (2). A 250 mL Erlenmeyer flask is equipped with a Teflon-coated magnetic stir bar (3.5 cm x 0.5 cm, rod-shaped). The flask is charged with (E)-di-tert-butyl(hex-2-en-1-yloxy) silanol (1) (7.03 g, 27.1 mmol, 1 equiv), using a 1 mL glass pipette, and CH2Cl2 (110 mL), using a 250 mL glass graduated cylinder. The reaction flask is then placed in an ice-water bath (Figure 5A). m-chloroperoxybenzoic acid (9.40 g, ~37.9 mmol, ~1.4 equiv) (Note 20) is added to the flask portion-wise over 10 min. The flask is capped with aluminum foil (Figure 5B). The reaction mixture is stirred (500 rpm) for 5 min at 0 ℃, then the ice-water bath is removed. The reaction mixture is allowed to warm to room temperature (20 ℃) with stirring over a period of 3 h (Figure 5C). The progress of the reaction is monitored by TLC (Figure 6). After full consumption of the starting material (~3 h), the reaction is cooled to 0 ℃ using an ice-water bath and quenched by slowly adding saturated aqueous sodium thiosulfate solution (50 mL) (Note 21). The reaction mixture is then transferred to a 1 L separatory funnel. The reaction flask is rinsed with diethyl ether (75 mL), which is added to the separatory funnel. (Note 22) (Figure 7). The aqueous layer is separated, and the organic layer is washed with 1 M aqueous NaOH solution (3 x 75 mL) (Note 23). The organic layer is dried over anhydrous MgSO4 (5 g), filtered through a 60 mL sintered glass funnel (50 mm frit, coarse) under house vacuum (~75 mm Hg) into a 500 mL round-bottomed flask and concentrated under reduced pressure using a rotary evaporator (Note 12). The residue is purified by flash column chromatography (Figure 8) (Note 24) on silica gel. The fractions containing pure product are combined and concentrated under reduced pressure on a rotary evaporator (Note 17), followed by high vacuum (20 ℃, <1 mmHg) for 2 h to afford 2 (Figure 9) as a colorless oil (6.85 g, 24.9 mmol, 92%) (Notes 25 and 26).

Figure 5. (A) Reaction mixture cooled to 0 ℃ using an ice-water bath prior to the addition of mCPBA. (B) Directly after addition of mCPBA. (C) After warming to room temperature (20 ℃) , close to reaction completion. (Photos provided by checkers).
Figure 6. The progress of the reaction is monitored by TLC analysis on silica gel plates using 10% EtOAc in hexanes as the eluent. TLC spots are visualized by staining with ceric ammonium molybdate stain. Note: S = starting material, C = co-spot lane, R= reaction mixture; Compound 1 (Rf = 0.54), epoxide product 2 (Rf = 0.42) (Photo provided by checkers)
Figure 7. Reaction mixture after quenching with saturated aqueous Na2S2O3 solution and diluting with Et2O (Photo provided by checkers)
Figure 8. Pure compound 2 is found in fractions 45-89 (Photo provided by checkers)
Figure 9. Di-tert-butyl(((2R*,3R*)-3-propyloxiran-2-yl)methoxy)silanol (2) is a colorless oil (Photo provided by checkers)
C. (4S*,5R*)-2,2-Di-tert-butyl-4-propyl-1,3,2-dioxasilinan-5-ol (3). An oven-dried, single-necked (24/40 joint) 500 mL round-bottomed flask is equipped with a Teflon-coated magnetic stir bar (4.0 cm x 1.0 cm, oval) and septum, and purged for 30 min with argon via a 20G inlet from a Schlenk line and a 20G exit needle connected to an oil bubbler (Figure 1A). The flask is then charged with di-tert-butyl(((2R*,3R*)-3-propyloxiran-2-yl)methoxy)-silanol (2) (5.84 g, 21.3 mmol, 1 equiv), and CH2Cl2 (200 mL) using a plastic syringe fitted with 20G needle (Note 4). Sodium bicarbonate (1.85 g, 22.0 mmol, 1 equiv) (Note 27) is added in one portion (Figure 10A). Stirring (250 rpm) is commenced. The reaction flask is then placed in an ice-water bath (Figure 10B). After 10 min, Ph3CBF4 (1.06 g, 3.21 mmol, 0.15 equiv) (Note 28) is added in three approximately equal portions over 5 min (Figure 10C).

Figure 10. (A) After addition of NaHCO3. (B) The reaction is cooled to 0 ℃ prior to addition of Ph3CBF4. (C) After addition of Ph3CBF4 (at 0 ℃), the reaction mixture turns bright yellow, and this color persists even after warming to room temperature (20 ℃) (Photos provided by checkers)
The ice-water bath is removed, and the reaction mixture is allowed to warm to room temperature (20 ℃). The progress of the reaction is monitored by TLC (Figure 11). After the consumption of starting material (~ 2 h after addition of Ph3CBF4), the reaction is cooled again using an ice-water bath. The reaction is quenched by adding saturated aqueous NaHCO3 solution (50 mL) (Note 9) and further diluted with an additional 20 mL of CH2Cl2 (Figure 12A). The reaction mixture is transferred to a 1 L separatory funnel (Figure 12B). The aqueous layer is removed, and the organic layer is washed with saturated aqueous NaHCO3 solution (2 x 50 mL). The organic layer is dried over anhydrous MgSO4 (5 g) and filtered through a 60 mL sintered glass funnel (50 mm frit, coarse) under house vacuum (~75 mmHg) into a 500 mL round-bottomed flask. After concentrating under reduced pressure on a rotary evaporator (Note 12), the residue is purified by flash column chromatography on silica gel (Notes 29 and 30). The fractions containing pure product 3 are combined (Figure 13) and concentrated under reduced pressure on a rotary evaporator (Note 12) followed by high vacuum (20 ℃, <1 mmHg) for 2 h to give 3 as a white solid (Figure 14A) (4.85 g, 17.7 mmol, 83%) (Notes 31 and 32).
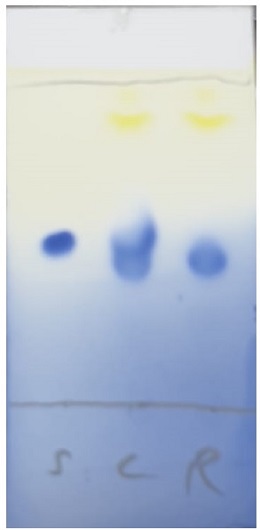
Figure 11. The progress of the reaction is monitored by TLC analysis on silica gel with 2% acetone in CH2Cl2 as the eluent (Note 25). Spots are visualized after the plate is dipped in ceric ammonium molybdate stain and heated. Note: S = starting material (2), C = co-spot lane, R = reaction mixture containing catalyst (Rf = 0.86, yellow spot) and product 3. Compound 2 (Rf = 0.46), Compound 3 (Rf = 0.44) (Photo provided by checkers).
Figure 12. (A) Reaction mixture becomes pale yellow after quenching with saturated aqueous NaHCO3 solution. (B) After partitioning aqueous and organic layers in a 1 L separatory funnel (photos provided by submitters)
Figure 13. Pure compound 3 is found in fractions 56-82 (Photo provided by checkers)
Figure 14. (A) (4S*,5R*)-2,2-Di-tert-butyl-4-propyl-1,3,2-dioxasilinan-5-ol (3) is a white solid after purification. (B) Crystals suitable for X-ray diffraction analysis are grown by slow evaporation from hexanes (photos provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
di-tert-butylsilyl bis(trifluoromethanesulfonate),
m-chloroperoxybenzoic acid,
diethyl ether,
acetone,
toluene,
CH2Cl2,
trans-2-hexen-1-ol,
2,6-lutidine, concentrated
hydrochloric acid,
magnesium sulfate, silica gel,
hexanes,
acetone, concentrated
sulfuric acid,
sodium bicarbonate,
sodium thiosulfate pentahydrate,
sodium hydroxide,
ammonium molybdate, and
ceric ammonium molybdate.
2. The submitters used a nitrogen balloon instead of an argon line.
3.
Di-tert-butylsilyl bis(trifluoromethanesulfonate) was purchased from Combi-Blocks (96% purity), which was used as received. The checkers purchased
di-tert-butylsilyl bis(trifluoromethanesulfonate) from Shanghai Macklin Biochemical Technology Co., Ltd (97% purity), which was used as received.
4.
CH2Cl2 was purchased from Sigma-Aldrich (≥99.5%, ACS reagent grade) and was used as received. The checkers purchased
CH2Cl2 from Anaqua Global International Inc. Ltd (≥99.5%, ACS reagent grade) which was used as received.
5.
2,6-Lutidine was purchased from TCI America (purity > 98 %) and was used as received. The checkers purchased
2,6-lutidine from TCI China (purity > 98 %) which was used as received.
6.
Trans-2-hexen-1-ol was purchased from TCI America (purity > 95 %) and used as received. The checkers purchased
trans-2-hexen-1-ol from TCI China (purity > 95 %) which was used as received.
7. Thin layer chromatography (TLC) was performed on silica gel 60 F254 (aluminum TLC plates). The progress of the reaction is monitored by TLC analysis eluting with 10%
EtOAc in
hexanes. Spots are visualized after the plate is dipped in
ceric ammonium molybdate stain and heated.
8.
Ceric ammonium molybdate stain was prepared by dissolving 12 g of
ammonium molybdate, 0.5 g of
ceric ammonium molybdate, and 15 mL of concentrated
H2SO4 in 235 mL water. The checkers prepared
ceric ammonium molybdate stain by dissolving 5 g of
ammonium molybdate, 1 g of cerium sulfate, and 10 mL of concentrated
H2SO4 in 90 mL water.
9. The checkers purchased
sodium bicarbonate (AR grade) from Dieckmann (Hong Kong) Chemical Industry Co. Ltd., which was used as received.
10.
Hydrochloric acid (Certified ACS Plus, 36.5 to 38%) was purchased from Fisher Chemical. A 1 M
HCl aqueous solution was prepared by adding 83 mL of th4 concentrated
HCl to 917 mL of deionized water. The checkers purchased
hydrochloric acid (AR grade, 37%) from RCI Labscan Ltd.
11.
Magnesium sulfate (anhydrous) was purchased from Fisher Scientific and used directly. The checkers purchased
magnesium sulfate (AR grade, anhydrous) from Dieckmann (Hong Kong) Chemical Industry Co. Ltd., which was used directly.
12. The checkers used a Heidolph Hei-VAP Core Rotovap with a bath temperature of 25 ℃ and at a vacuum of 200 mmHg.
13. The checkers purchased ethyl acetate from Duksan Pure Chemicals Co. Ltd. (GR grade), which was used as received.
14.
Hexanes (>98.5%) was purchased from Fisher Scientific and used as received. The checkers purchased
hexanes (ACS grade, >99.5%) from SK Chemicals Co. Ltd., which was used as received.
15. Silica Gel (grade 60, 230-400 mesh) was purchased from Fisher Scientific. The checkers purchased silica gel (0.040-0.063 mm) from Merck KGaA, which was used as received.
16. For flash column chromatography, a 30 cm (L) x 5 cm (W) glass column is charged with 170 g of silica gel and is flushed with 750 mL of 2%
EtOAc in
hexanes. The height of the silica gel is 16 cm. The crude product is dissolved in 20 mL of 2%
EtOAc in
hexanes and loaded onto the column. After full adsorption, the top of the silica gel is layered with sand (2 cm). The column was eluted using 500 mL of 2%
EtOAc in
hexanes, 500 mL of 4%
EtOAc in
hexanes, then 1 L of 6%
EtOAc in
hexanes. Fractions of 20 mL were collected. Fractions were checked using TLC (10%
EtOAc in
hexanes) and visualized by
ceric ammonium molybdate stain.
17. The checkers used a Heidolph Hei-VAP Core Rotovap with a bath temperature of 30 ℃ and a vacuum at 100 mmHg.
18. Analytical data for
(E)-di-tert-butyl(hex-2-en-1-yloxy)silanol (
1):
1H NMR
pdf (500 MHz, CDCl
3) δ: 5.67 (dtt,
J = 14.7, 6.6, 1.4 Hz, 1H), 5.56 (dtt,
J = 15.3, 5.3, 1.3 Hz, 1H), 4.29 (dq,
J = 5.3, 1.2 Hz, 2H), 2.07 - 1.96 (m, 2H), 1.85 (s, 1H) 1.40 (h,
J = 7.4 Hz, 2H), 1.02 (s, 18H), 0.90 (t,
J = 7.4 Hz, 3H);
13C{
1H} NMR
pdf (101 MHz, CDCl
3) δ: 131.3, 129.6, 64.3, 34.4, 27.6, 22.5, 20.6, 13.8; IR (ATR) ν 3418, 2962, 2933, 2859, 1683, 1470, 138, 1364, 1104, 1062, 1012, 969, 826, 648, 442 cm
-1. HRMS (ESI)
m/z = [M+ Na
+] calculated for C
14H
30O
2Si 281.1913, Found 281.1915. The purity of
1 was determined to be 99.0% by qNMR
pdf using
4-nitrotoluene (purity 99.5%, purchased from Shanghai Aladdin Biochemical Technology Co., Ltd.) as the internal standard.
19. A second reaction performed by the checkers on half scale provided 4.43 g (67% yield) of compound
1.
20.
m-chloroperoxybenzoic acid (70-75% purity, with the balance being 3-chlorobenzoic acid and water) was purchased from Thermo Scientific and was used without purification.
21.
Sodium thiosulfate pentahydrate (>99%) was purchased from Fisher Scientific and used as received. The checkers purchased
sodium thiosulfate pentahydrate (> 99%) from TCI China which was used as received.
22.
Diethyl ether (Laboratory grade) was purchased from Fisher Scientific and used as received. The checkers purchased
Et2O (AR grade) from RCI Labscan Ltd. and used the solvent as received.
23.
Sodium hydroxide pellets were purchased from Acros Organics. A 1 M
NaOH solution was prepared by dissolving 20 g of
NaOH pellets in 500 mL deionized water. The checkers purchased
sodium hydroxide pellets (AR grade) from Dieckmann (Hong Kong) Chemical Industry Co. Ltd., which was used as received.
24. For flash column chromatography, a 30 cm (L) x 5 cm (W) glass column is charged with 160 g of silica gel and is flushed with 500 mL of 95:5
hexanes/
EtOAc. The height of the packed silica gel is 15 cm. The crude product is dissolved in 15 mL of 5%
EtOAc in
hexanes and loaded onto the column. After full adsorption, the top of the silica gel is layered with sand (2 cm). The column is eluted with 5%
EtOAc in
hexanes (500 mL), 12%
EtOAcc in
hexanes (500 mL), then 15%
EtOAc in
hexanes (250 mL). Fraction of 20 mL were collected. Fractions were checked using TLC (10%
EtOAc in
hexanes) (Figure 8) and visualized by
ceric ammonium molybdate stain.
25. Analytical data for
di-tert-Butyl(((2R*,3R*)-3-propyloxiran-2-yl)methoxy)silanol (
2):
1H NMR
pdf (400 MHz, CDCl
3) δ: 4.12 (ddd,
J = 12.4, 2.4, 1.1 Hz, 1H), 3.71 (dd
, J = 12.3, 5.8 Hz, 1H), 2.93 (tq,
J = 4.7, 2.4 Hz, 2H), 1.61 - 1.37 (m, 4H), 1.03 (s, 9H), 1.01 (s, 9H), 0.95 (t,
J = 7.2 Hz, 3H);
13C{
1H} NMR
pdf (101 MHz, CDCl
3) δ: 64.1, 59.3, 57.0, 33.7, 27.5, 20.74, 20.67, 19.4, 14.0; IR (ATR) ν 3419, 2963, 2933, 2859, 1471, 1387, 1364, 1242, 1129, 1095, 1012, 939, 851, 827, 772, 649, 442 cm
-1.; HRMS (ESI)
m/z = [M + Na
+] calculated for C
14H
30O
3Si 297.1862, Found 297.1865. The purity of
2 was determined to be 99.5% by qNMR
pdf using
4-nitrotoluene (purity 99.5%, purchased from Shanghai Aladdin Biochemical Technology Co., Ltd.) as the internal standard.
26. A second reaction performed by the checkers on half scale provided 3.48 g (94%) of compound
2.27.
Sodium bicarbonate (ACS reagent grade) was purchased from Oakwood Chemical and used as received. The checkers purchased
sodium bicarbonate (AR grade) from Dieckmann (Hong Kong) Chemical Industry Co. Ltd., which was used as received.
28.
Triphenylcarbenium tetrafluoroborate (
Ph3CBF4) was purchased from Alfa Aesar (97%) and used as received. The checkers purchased
triphenylcarbenium tetrafluoroborate (
Ph3CBF4) from TCI China (98%) which was used as received.
29. For flash column chromatography, a 30 cm (L) x 5 cm (W) glass column is charged with 160 g of silica gel and is flushed with 500 mL of
CH2Cl2. The height of the silica gel is 15 cm. The crude product is dissolved in 10 mL of
CH2Cl2 and loaded onto the column. After full adsorption, the top of the silica gel is layered with sand (2 cm). The column is eluted with
CH2Cl2 (500 mL), then 2%
acetone in
CH2Cl2 (500 mL) (
Note 30), 4%
acetone in
CH2Cl2 (250 mL), and 6%
acetone in
CH2Cl2 (500 mL). Fractions of 20 mL were collected. Fractions were checked using thin-layer chromatography (2%
acetone in
CH2Cl2) and visualized using
ceric ammonium molybdate stain (Figure 13).
30.
Acetone (Tech Grade) was purchased from Dah Fat Chemical International Ltd. and used as received.
31. Analytical data for (
4S*,5R*)-2,2-di-tert-butyl-4-propyl-1,3,2-dioxasilinan-5-ol:
1H NMR
pdf (400 MHz, CDCl
3) δ: 4.08 (dd,
J= 10.5, 4.6 Hz, 1H), 3.84 - 3.72 (m, 2H), 3.53 (td,
J = 9.3, 4.5 Hz, 1H), 1.83 - 1.70 (m, 1H), 1.67 - 1.32 (m, 4H), 1.04 (s, 9H), 1.00 - 0.93 (m, 12H);
13C{
1H} NMR
pdf (101 MHz, CDCl
3) δ: 78.1, 70.8, 68.8, 36.8, 27.7, 27.2, 22.8, 20.2, 18.1, 14.2. IR (ATR) ν 3409, 2960, 2934, 2861, 1471, 1387, 1365, 1236, 1215, 1155, 1117, 1080, 1065, 1053, 1040, 1025, 1010, 1000, 938, 895, 825, 787, 769, 756, 727, 651, 598, 460, 440 cm
-1; HRMS (ESI)
m/z = [M + Na
+] calculated for C
14H
30O
3Si 297.1862, Found 297.1861. mp 67-69 ℃. The purity of
3 was determined to be 97.3% by qNMR
pdf using
4-nitrotoluene (purity 99.5%, purchased from Shanghai Aladdin Biochemical Technology Co., Ltd.) as the internal standard. Single crystals of
3 suitable for X-ray diffraction analysis were grown by dissolving 1 gram of
3 (obtained from chromatographic purification) in 5 mL of
hexanes in a 25 mL round-bottomed flask (Figure 14B). This mixture was heated in a 60 ℃ oil bath for one min, then cooled to room temperature (20 ℃ ) over 5 min. The flask was sealed with a rubber septum and kept in the refrigerator (~4 ℃) for two days. The septum was pierced with two 20G vent needles and left on the benchtop at ambient temperature for one day. Over this time, needle-like, colorless crystals formed in the flask. X-ray diffraction analysis of one of these crystals (CCDC 2245378) unambiguously confirmed the identity and relative stereochemistry of
3.
32. A second reaction performed by the checkers on half scale provided 2.45 g (84%) of compound
3.
3. Discussion
Table 1. Substrate scope with alkyl epoxides
Table 1 (continued). Substrate scope with alkyl epoxides
Table 2. Substrate scope with BINOL-phosphoric acid conditions
Scheme 1. A short preparation of protected D-arabitol
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved