Org. Synth. 2024, 101, 61-80
DOI: 10.15227/orgsyn.101.0061
Preparation of Radical Clocks Bearing Carbonyl Groups: Synthesis of N-Methoxy-N-methylspiro[cyclopropane-1,9'-fluorene]-2-carboxamide
Submitted by Nicole D. Bartolo, Ryan N. Robson, Collin H. Witt, and K. A. Woerpel*
1Checked by Kevin Lee and Dirk Trauner
1. Procedure (Note 1)
A. (9H-Fluoren-9-ylidene)hydrazine (2). A 1-L, three-necked, round-bottomed flask equipped with a Teflon-coated magnetic stir bar (football-shaped, approximately 7 cm), one glass stopper, and a thermometer. The one remaining open neck (Figure 1A) is charged with 9-fluorenone (1, 50.0 g, 278 mmol, 1.0 equiv) (Note 2) and EtOH (500 mL) (Note 3). The solution is stirred at ≈900 rpm (Note 4) and heated to an internal temperature of 40 ℃ (Note 5) for 45 min until a homogenous solution is created (Figure 1A). Hydrazine monohydrate (50.0 mL, 789 mmol, 2.8 equiv) (Note 6) is then added in a single portion using a graduated cylinder through the open neck of the flask. The open neck of the flask is equipped with a reflux condenser, and the oil bath temperature is raised to 105 ℃ and maintained at this temperature for an additional 3.5 h (Note 7) (Figure 1B), during which time the progress of the reaction is monitored by TLC (Figure 2) (Note 8). The oil bath is removed and stirring of the solution is maintained at ≈900 rpm. The reaction is allowed to cool for 1 h to an internal temperature of 20 ℃, which facilitates the precipitation of (9H-fluoren-9-ylidene)hydrazine (2) from the solution (Figure 1C) (Note 9). The resulting crystals are collected by vacuum filtration on an 11-cm Büchner funnel equipped with filter paper (Figure 3). The crystals are dried under a flow of air provided by the vacuum for 2 h, which is followed by additional drying under high vacuum (3 mmHg) for 4 h to afford (9H-fluoren-9-ylidene)hydrazine (2) as yellow needles (48.1 g, 90%) (Notes 10, 11, and 12). The crystals were used in the next step without further purification (Note 13).

Figure 1. A) Formation of hydrazone at 40 ℃ prior to dissolution; B) Reaction mixture following completion of hydrazine addition; C) Crystallization upon cooling with continuous stirring
Figure 2. TLC with 10%vol ethyl acetate/hexanes; A) observed with 254 nm light eluted; B) after staining with vanillin
Figure 3. Crystallization of hydrazone 2 A) Prior to filtration; B) Wet crystals after filtration; C) Dried crystals. The pH paper is included for color reference
B. Methyl spiro[cyclopropane-1,9'-fluorene]-2-carboxylate (4). A 500-mL, three-necked, round-bottomed flask equipped with a solid addition funnel (50 mL), a mechanical stirrer (Note 14), and a thermometer is charged with (9H-fluoren-9-ylidene)hydrazine (2, 46.2 g, 220 mmol, 1.0 equiv) (Note 15), nickel(II) hydroxide (6.60 g, 0.3 equiv) (Note 16), and methyl acrylate (3) (54.0 mL, 51.3 g, 2.5 equiv) (Notes 17 and 18) (Figure 4). The mixture is stirred at 400 rpm and rapidly lowered into an oil bath heated to 80 ℃. After stirring for 45 min, a green colored solution is obtained (Figure 4B), and iodosobenzene (63.0 g, 1.2 equiv) (Note 19) is added via the solid addition funnel at a rate of 0.5 g per minute, maintaining an internal temperature of the reaction mixture below 85 ℃ (Notes 20 and 21). As the iodosobenzene is added, the reaction evolves nitrogen gas and the mixture progressively changes color from green (Figure 4C) to brown (Figure 4D) to a deep orange/red (Figure 4E). After addition of iodosobenzene is complete (approximately 2 h), the reaction mixture is stirred for an additional 75 min with an oil bath temperature of 80 ℃. The progress of the reaction is monitored by TLC (Figure 5). The reaction mixture is removed from the oil bath and allowed to cool over 45 min. The reaction mixture is then diluted with dichloromethane (100 mL) and vacuum filtered through Celite (65 g) placed in a fritted glass funnel (9 cm diameter). The Celite is washed (Note 22) with four 250-mL portions of dichloromethane, and the filtrate is concentrated on a rotary evaporator (75 mmHg) with the water bath set at 40 ℃. The resulting oil is further dried under high vacuum (3 mmHg) for 2 h to afford a red oil, which solidifies upon standing at 20 ℃ for 18 h (Figure 6A). The red solid is suspended in dichloromethane (50 mL) and charged on a fritted glass funnel (13 cm diameter) filled with silica (400 g). Hexanes (500 mL) is forced through the silica with vacuum, and the filtrate is discarded. The silica is then eluted with 5:95 ethyl acetate: hexanes (3 L) using vacuum, and the resulting filtrate is concentrated by rotary evaporator (75 mmHg) with the water bath set to 40 ℃. The resulting orange solid is then further dried by high vacuum (3 mmHg) for 2.5 h to afford methyl spiro[cyclopropane-1,9'-fluorene]-2-carboxylate (4) (55.45 g, 84%) (Notes 23, 24, and 25) as an orange solid, which is used in the next step without further purification (Figure 6B) (Note 26).

Figure 4. A) Setup of carbene addition reaction; B) Reaction prior to addition; C) Reaction at beginning of addition; D) Reaction approximately halfway through addition; E) Reaction at completion
Figure 5. TLC for carbene addition reaction observed with A) 254 nm light in 10% ethyl acetate/hexanes; B) After staining with vanillin
Figure 6. A) Red solid that forms upon standing of 4 at 20 ℃ for 18 h; B) Dried filtrate from elution with 5:95 ethyl acetate:hexanes
C. N-Methoxy-N-methylspiro[cyclopropane-1,9'-fluorene]-2-carboxamide (5). A 3-L, three-necked, round-bottomed flask (Figure 7A) (Note 27), equipped with a pressure equalizing dropping funnel capped with a rubber septum with a nitrogen inlet needle and mechanical stirrer, is charged with methyl spiro[cyclopropane-1,9'-fluorene]-2-carboxylate (4) (50.1 g, 200mmol, 1.0 equiv) and N,O-dimethylhydroxylamine (30.4 g, 1.55 equiv) (Note 28). A low temperature thermometer is attached to the third neck and a vent needle (18 gauge) is added to the rubber septum. The flask is purged with a stream of nitrogen through the inlet needle for 10 min, after which the vent needle is removed, and an inert atmosphere is maintained for the remainder of the reaction. Tetrahydrofuran (THF) (400 mL) (Note 29) is then added to the flask using a gas-tight syringe, and the reaction mixture is stirred at 400 rpm. The flask is cooled to an internal temperature of -20 ℃ using a chiller with the external temperature set at -50 ℃ (Notes 30 and 31). The pressure equalizing addition funnel is charged with isopropylmagnesium chloride (300 mL, 2 M in THF, 3.0 equiv) (Note 32), which is then added dropwise to the reaction mixture over 3.5 h while maintaining an internal temperature between -20 ℃ and -15 ℃ (Figure 7B). After the addition of isopropylmagnesium chloride is complete, the flask is removed from the chiller and the reaction mixture is allowed to warm for 2.5 h to an internal temperature of 8 ℃. The excess Grignard reagent is consumed by addition of aq. HCl (500 mL, 2.0 M) added as a single portion (Note 33). The reaction mixture is transferred to a 2 L separatory funnel and is extracted with ethyl acetate (3 x 300 mL) (Note 34).

Figure 7. A) Reaction setup for Weinreb amide formation; B) Reaction mixture during addition of isopropylmagnesium chloride
The combined organic layers are washed with saturated aq. NaCl solution (500 mL), dried over MgSO4 (15 g), filtered through cotton, and concentrated by rotary evaporation (75 mmHg with 30 ℃ external water bath temperature) followed by high vacuum (3 mmHg, 8 h) to afford a yellow solid (Figure 8A) (Note 35). A slurry is prepared from silica (400g) (Note 36) using a mixture of 15:85 ethyl acetate and hexane (1L) and is loaded into a glass column (11 x 14 cm). The column is then packed with pressurized air and a layer of sand is added (60g). The product is then dissolved in CH2Cl2 (25 mL) and eluted with 500 mL of 15% EtOAc-hexanes (15:85 EtOAc: hexanes), during which a yellow band of impurity elutes and fraction collection (20-mL fractions in 16×150 mm test tubes) is initiated. Elution continues with 5 L of 15% EtOAc-hexanes (15:85 EtOAc:hexanes, Figure 9). The desired product is obtained in fractions 65-216, which are concentrated by rotary evaporation (75 mmHg with 35 ℃ external water bath temperature) then dried under high vacuum (3 mmHg, 8 h) to afford a pale-yellow solid (34.7 g, 55% yield) (Notes 37, 38, and 39) (Figure 8B).

Figure 8. A) Yellow solid of the unpurified 5 suitable for further reactions; B) The purified white solid
Figure 9. TLC of column fractions observed under 254 nm UV irradiation gradients are for every sixth fraction
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011); the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
9-fluorenone,
hydrazine,
ethanol,
sodium hydroxide,
N,O-dimethyl hydroxylamine,
isopropyl magnesium chloride,
tetrahydrofuran, as well as the proper procedures for flame drying and cryogenic reactions. Special care should be taken with the carbene addition step as it is highly exothermic and produces
N2 gas.
2. The authors purchased
9-fluorenone (98%) from Oakwood Chemical and used as received. The checkers purchased
9-fluorenone (98%) from Acros Organics and used as received.
3.
Ethanol - denatured was obtained from Fisher Scientific and used as received by the authors. The checkers purchased 200 proof
ethanol from Decon Labs and used it as received.
4. Magnetic stirring was performed using an IKA RCT basic stirrer.
5. Heating was performed using an IKA RCT basic heater with a probe for maintaining temperature with a silicon oil bath. The oil bath temperature was set to 50 ℃.
6.
Hydrazine monohydrate (79%) was obtained from Tokyo Chemical Institute and used as received.
7. During this stirring period, some
(9H-fluoren-9-ylidene)hydrazine (
2) may precipitate out of solution. This precipitation does not affect the outcome of the reaction.
8. Reaction progress can be monitored by TLC using 10%
vol EtOAc-
hexanes. The plate should be visualized with both UV light and staining with vanillin due to the small change in
Rf (Figure 2).
9. Rapid stirring ensures that fine needle crystals are formed and prevents clumping of the crystals. Fine yellow needles will form in an orange solution.
10.
(9H-Fluoren-9-ylidene)hydrazine (
2): mp = 150.2-150.6 ℃;
1H NMR
pdf (600 MHz, CDCl
3) δ: 7.92 (d,
J = 7.6 Hz, 1H), 7.79 (t,
J = 7.8 Hz, 2H), 7.68 (d,
J = 7.7 Hz, 1H), 7.46 (m, 1H), 7.37 (m, 2H), 7.33 (m, 1H), 6.42 (br s, 2H);
13C NMR
pdf (150 MHz, CDCl
3) δ: 145.7(C), 141.4(C), 138.6 (C), 137.8 (C), 130.3 (C), 129.8 (CH), 128.6 (CH), 128.0 (CH), 127.8 (CH), 125.6 (CH), 120.8 (CH), 120.6 (CH), 119.6 (CH); IR (thin film) 3389, 3316, 3202, 3090, 3073, 3062, 3058, 3034, 2900, 1640, 1614, 1580, 1480, 1448, 1351, 1334, 1302, 1268, 1220, 1998, 1166, 1156, 1114, 1068, 1034, 1005, 981, 972, 936, 868, 861, 779, 762, 730, 667, 641, 619, 552, 497, 446, 418 cm
-1. HRMS (EI)
m/z [M+] calcd for C
13H
10N
2: 194.0844. Found: 194.0854.
11. Purity was determined to be >95% through quantitative NMR
pdf using mesitylene (98%, Oakwood Chemical) as an internal standard.
12. A second reaction on the same scale performed by the checkers provided 46.52 g (87%) of the product with 93% purity.
13. Attempts at further purification by recrystallization from
ethanol led to a significant decrease in product yield. As such, the material was used without additional purification
14. Mechanical stirring was performed using a VELP Scientifica overhead stirrer (part number: F201A0152).
15. In lieu of performing step A, hydrazone
4 can be purchased in 97% purity from Fisher Scientific.
16. Nickel (II) hydroxide (61% Nickel) was obtained by the authors from Alfa Aesar and used as received. The checkers purchased nickel (II) hydroxide) from Acros Organics and used as received.
17. Methyl acrylate (99%) stabilized was obtained by the authors and checkers from Acros Organics and used as received.
18. The use of excess alkene in these types of Ni-catalyzed cyclopropanation reactions is required to minimize side reactions of the hydrazone starting material. It was determined through experimentation that the amount of methyl acrylate could be reduced from the usual 6.4 equivalents
2 to 2.5 equivalents without a significant reduction in yield.
19.
iodosobenzene is prepared according to
Org. Synth. 1963, 43, 60, but instead of trituration the wet solid is washed with chloroform before drying. The pale-yellow solid is ground to a fine powder prior to use. For the preparation of
iodosobenzene,
sodium hydroxide - pellets (98%) were obtained from Oakwood Chemical and used as received. (Diacetoxyiodo)benzene (98%) was obtained from Oakwood Chemical and used as received.
20. CAUTION: Initial addition of
iodosobenzene generates a violent exotherm and should be performed with caution and in small quantities until the exotherm subsides.
21. Condensation at the neck of the addition funnel may result in clumping of the
iodosobenzene that can lead to a blockage of the addition funnel. This blockage can be dislodged into the reaction mixture with a glass rod as needed.
22. The filter cake, which may contain unreacted
iodosobenzene, was collected and stored in a properly labeled solid waste container.
23.
Methyl spiro[cyclopropane-1,9'-fluorene]-2-carboxylate (
4):
1H NMR
pdf (600 MHz, CDCl
3) δ: 7.82-7.79 (m, 1H), 7.58 (d,
J = 7.7, 1H), 7.40-7.36 (m, 2H), 7.32-7.26 (m, 2H), 7.04 (d,
J = 7.5, 1H), 3.63 (s, 3H), 2.77-2.73 (m, 1H), 2.47-2.44 (dd,
J = 7.5, 5.2, 1H), 2.16-2.12 (dd,
J = 8.4, 5.2, 1H);
13C NMR
pdf (150 MHz, CDCl
3) δ: 170.0 (C), 146.4 (C), 142.5 (C), 141.0 (C), 139.8 (C), 127.2 (CH), 127.0 (CH), 126.9 (CH), 122.9 (CH), 119.90 (CH), 119.87 (CH), 118.7 (CH), 52.0 (CH
3), 37.1 (C), 32.7 (CH), 20.9 (CH
2). IR (film) 3037, 2955, 1728, 1478, 1201, 937 cm
-1; HRMS (EI) [M + H] calcd for C
17H
14O
2: 250.2970. Found: 250.1007.
24. Purity was determined to be 65% through quantitative NMR
pdf using mesitylene (98%, Oakwood Chemical) as an internal standard.
25. A second reaction on the same scale performed by the checkers provided 53.52 g (83%) of the product with 67% purity.
26. On a smaller scale synthesis,
2 methyl ester 4 was purified by flash chromatography using 5%
EtOAc-
hexanes as the eluent.
27. All glassware was flame-dried and cooled under high vacuum (4 mbar) and purged with a nitrogen atmosphere. Alternatively, the glassware can be oven dried (110 ℃) overnight and cooled under high vacuum before use.
28.
N,O-Dimethyl hydroxylamine hydrochloride (98%) was obtained from Oakwood Chemical and used as received.
29.
THF (inhibitor-free HPLC ≥ 99.9%) was obtained by the authors from Sigma-Aldrich and passed through a puresolv solvent purification system. The checkers obtained
THF (inhibitor-free HPLC ≥ 99.9%) from Fisher Scientific and passed through activated alumina using a JC Meyer solvent purification system.
30. Cryogenic cooling was performed with a Julabo FT-902 immersion cooler in an
acetone bath.
31. A low external temperature is used in this step to counteract the resulting exotherm that occurs during addition of the isopropylmagnesium chloride to the reaction mixture.
32.
Isopropyl magnesium chloride [2 M] in
THF was obtained from Sigma Aldrich and used as received.
33. The reaction mixture rose to an internal temperature of 39 ℃ during the addition of aq.
HCl.
34. Any minor emulsion from the first extraction is carried through to the second extraction where it resolved into two distinct layers.
35. If desired, this yellow solid
N-methoxy-N-methylspiro[cyclopropane-1,9'-fluorene]-2-carboxamide can be used without further purification. The authors report that addition of phenylmagnesium chloride to the crude
5 (82% purity) resulted in a yield of 93% of the corresponding phenyl ketone.
36. Silica was Irregular Silica Gel, P60 40-63 μm, 60 Å was obtained from SiliaFlash.
37.
N-Methoxy-N-methylspiro[cyclopropane-1,9'-fluorene]-2-carboxamide (
5). mp = 80.2-81.5 ℃;
1H NMR
pdf (600 MHz, CDCl
3) δ: 7.81-7.79 (m, 2H), 7.46-7.44 (m, 1H), 7.40-7.25 (m, 4H), 7.12 (d,
J = 7.3, 1H), 3.24-3.19 (m, 1H), 3.12 (br s, 3H), 2.91 (br s, 3H), 2.56-2.53 (dd,
J = 7.7, 5.2, 1H), 2.10-2.07 (dd,
J = 8.4, 5.2, 1H);
13C NMR
pdf (150 MHz, CDCl
3) δ: 170.0 (C), 146.4 (C), 142.5 (C), 141.0 (C), 139.8 (C), 127.2 (CH), 127.0 (CH), 126.9 (CH), 122.9 (CH), 119.90 (CH), 119.87 (CH), 118.7 (CH), 52.0 (CH
3), 37.1 (C), 32.7 (CH), 20.9 (CH
2). IR (film): 2934, 1660, 1445, 1079, 909 cm
-1. HRMS (EI) [M + 1] calcd for C
18H
18NO
2: 280.1332. Found: 280.1338.
38. Purity was determined to be 99% through quantitative NMR
pdf using mesitylene (98%, Oakwood Chemical) as an internal standard.
39. A second reaction on the same scale performed by the checkers provided 37.8 g (60%) of the product with 99% purity.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Carbonyl compounds bearing radical clocks are useful tools for investigating the possibility of single-electron transfer in reaction mechanisms.
2,3,4 Compared to the use of radical traps to probe reaction mechanism, compounds bearing radical clocks can mimic more closely the standard reaction conditions of an organic transformation, and they are operationally simpler to use.
2,3,4,5,6,7,8,9,10,11,12,13,14 The presence of ring-opened radical coupling products in the reaction mixture indicates that a single-electron transfer step has occurred (Scheme 1).
2Scheme 1. Possible ring-opened products formed in the course of a reaction that undergoes single-electron transfer
The procedure described in this work provides a simple route to prepare a Weinreb amide bearing a 9-fluorenyl radical clock. The 9-fluorenyl radical clock was chosen because it opens an order of magnitude faster than the more commonly used 2,2-diphenylcyclopropylcarbinyl radical clock.
11 The 9-fluorenyl radical clock has not been used extensively due to difficulties handling the synthetic intermediates, which are prone to ring-opening.
14,15 The method reported here, compared to other methods of synthesizing similar radical clock probes,
3 limits the handling of a potentially reactive diazo compound,
16 instead using that intermediate directly to generate the requisite cyclopropane.
The Weinreb amide
17 (
5) was synthesized because it can serve as a single synthetic intermediate to generate a series of electronically and sterically differentiated carbonyl compounds. Because reaction mechanism may depend upon both the nature of the carbonyl group and the nature of the reagent being explored, it is important in some cases to test a reaction with multiple differentiated radical clocks.
2,18,19,20 Grignard reagents can be added to Weinreb amide
5 to synthesize various ketones bearing the 9-fluorenyl radical clock (Table 1,
6a-
6c). Additionally, an aldehyde bearing a 9-fluorenyl radical clock (
10) can be synthesized by reaction of the Weinreb amide with diisobutylaluminum hydride (Scheme 2).
Table 1. Grignard additions to Weinreb amide 5 *(when performed on unpurified 5)
Scheme 2. Reaction of diisobutylaluminum hydride (DIBAL-H) with Weinreb amide 3
The resulting carbonyl compounds synthesized through the Weinreb amide can be used to explore the possibility of single-electron transfer in a variety of synthetic transformations. Our investigations into the mechanisms through which Grignard reagents add to carbonyl compounds demonstrates the importance of exploring electronically and sterically differentiated carbonyl groups.
2 We note that ketones
6a-
6c were formed without evidence of any ring-opened products, which suggests that the reactions shown in Table 1 do not proceed by single-electron transfer mechanisms. Similarly, the addition of allylmagnesium chloride to radical clocks bearing a series of carbonyl compounds appear to proceed through a concerted, two-electron mechanism as demonstrated by the lack of ring-opened products in the reaction mixture (Scheme 3). Conversely, the mechanism through which additions of
tert- butylmagnesium chloride add to the same series of radical clocks demonstrated that reaction mechanism can be dependent on the electronic nature of the carbonyl group (Scheme 4). These substrates have also been used to explore the reaction mechanisms by which organolithium reagents and organocuprates add to carbonyl compounds.
2
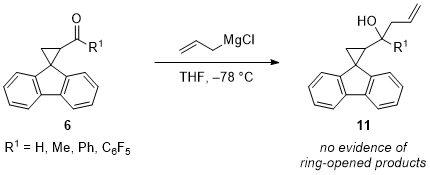
Scheme 3. Additions of allylmagnesium chloride to radical clocks bearing different carbonyl compounds suggest the reactions proceed through a two-electron mechanism
Scheme 4. Additions of tert-butylmagnesium chloride
Overall, the procedure reported here provides a simple route to synthesize carbonyl compounds bearing the 9-fluorenyl radical clock through a Weinreb amide. Compared to previous methods, no ring-opened products are detected in the course of the synthetic route. These compounds should provide a synthetically simple way to probe reaction mechanisms in the transformations of carbonyl groups and related functional groups.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
9-Fluorenone; (1) (486-25-9)
Hydrazine monohydrate; (7803-57-8)
Nickel(II) hydroxide; (12054-48-7)
Methyl acrylate; (96-33-3)
N,O-Dimethylhydroxylamine; (6638-79-5)
Isopropylmagnesium chloride; (1068-55-9)

|
Professor Woerpel received his Ph.D. at Harvard University studying with Professor David Evans, and he worked with Professor Robert Bergman at the University of California, Berkeley as a postdoctoral researcher. He is currently the Margaret and Herman Sokol Professor of Medicinal Chemistry at New York University. |

|
Nicole Bartolo received her Ph.D. in Chemistry at New York University under the guidance of Professor Keith Woerpel. She then completed a postdoctoral fellowship with a dual appointment at Harvard Medical School and Massachusetts General Hospital with Dr. Jacob Hooker. She is currently a Senior Scientist at AstraZeneca in Waltham, MA. |

|
Ryan Robson completed his Ph.D. at Deakin University under supervision for Associate Professor Frederick Pfeffer. He continued research as a postdoctoral associate at New York University and is currently a postdoctoral fellow at Monash University. |

|
Collin Witt received a B.S. degree in chemistry from Boston College in 2018. He completed his Ph.D. at New York University with Professor Keith Woerpel, where he studied stereoselective reactions of seven-membered-ring reactive intermediates. He is currently a Research Scientist at the Kostas Research Institute at Northeastern University. |

|
Kevin Lee attended William Paterson University of New Jersey (United States), completing his B.S. in Chemistry and Medicinal Biochemistry in 2022 under the supervision of Yalan Xing. That same year, he joined the University of Pennsylvania to obtain his Masters in Chemical Science degree under the supervision of Dirk Trauner. His research focuses on the asymmetric total synthesis of complex natural products. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved